Vyondys 53
Les Informations Présentées Sur Ce Site Ne Constituent Pas Un Avis Médical. Nous Ne Vendons Rien. L'Exactitude De La Traduction N'Est Pas Garantie. Clause De Non-Responsabilité
Résumé
Qu'est-ce que Vyondys 53?
Vyondys 53 (Golodirsen) est un oligonucléotide antisens indiqué pour le traitement de Dystrophie musculaire de Duchenne (DMD) chez les patients qui ont une mutation confirmée du gène DMD qui se prête à l'exon 53 saut.
Quels sont les effets secondaires de Vyondys 53?
Vyondys 53
- urticaire
- difficulté à respirer
- gonflement de ton visage lèvres langue ou gorge
- démangeaison
- éruption cutanée
- cloques ou pelage de la peau
- Urine brune ou rouge rose
- urine mousseuse et
- gonflement dans votre visage les mains ou l'estomac
Obtenez de l'aide médicale immédiatement si vous présentez l'un des symptômes énumérés ci-dessus.
Les effets secondaires de Vyondys 53 incluent:
- mal de tête
- fièvre
- chute
- douleurs abdominales
- coulant ou nez encombré
- toux
- vomissement
- nausée
- Pain d'administration
- maux de dos
- diarrhée
- vertiges
- entorse ligamentaire
- contusion
- grippe
- douleur de la bouche et de la gorge
- abrasion de la peau
- infection de l'oreille
- saisonnier allergie
- fréquence cardiaque rapide
- réaction liée au site du cathéter
- constipation et
- fracture
Cherchez des soins médicaux ou appelez le 911 à la fois si vous avez les effets secondaires graves suivants:
- Des symptômes oculaires graves tels que la perte de vision soudaine floue du tunnel de vision de la vision de la vision des yeux ou de l'enflure ou de voir des halos autour des lumières;
- Symptômes cardiaques graves tels que les battements cardiaques rapides ou battants; flotter dans votre poitrine; essoufflement; Et des étourdissements soudains étourdissement ou s'évanouir;
- Maux de tête sévères Confusion Slurred Speech Bras ou Ligne Proulitude Trouble de la merde Perte de coordination Sentiment des muscles très rigides très rigides Fièvre élevée Propice ou des tremblements.
Ce document ne contient pas tous les effets secondaires possibles et d'autres peuvent survenir. Vérifiez auprès de votre médecin des informations supplémentaires sur les effets secondaires.
Dosage pour vyondys 53
La dose de Vyondys 53 est de 30 milligrammes par kilogramme une fois par semaine.
Vyondys 53 chez les enfants
Vyondys 53 est indiqué pour le traitement de la dystrophie musculaire de Duchenne (DMD) chez les patients qui ont une mutation confirmée du gène DMD qui se prête à l'exon 53 saut, y compris les patients pédiatriques.
Quelles substances ou suppléments de médicaments interagissent avec Vyondys 53?
Vyondys 53 peut interagir avec d'autres médicaments.
Dites à votre médecin tous les médicaments et suppléments que vous utilisez.
Vyondys 53 pendant la grossesse et l'allaitement
Dites à votre médecin si vous êtes enceinte ou prévoyez de devenir enceinte avant d'utiliser Vyondys 53; On ne sait pas comment cela affecterait un fœtus. On ne sait pas si Vyondys 53 passe dans le lait maternel. Consultez votre médecin avant l'allaitement.
Informations Complémentaires
Notre injection de Vyondys 53 (Golodirsen) pour un centre de médicaments à effet secondaire d'usage intraveineux offre une vue complète des informations de médicament disponibles sur les effets secondaires potentiels lors de la prise de ce médicament.
Ce n'est pas une liste complète des effets secondaires et d'autres peuvent survenir. Appelez votre médecin pour des conseils médicaux sur les effets secondaires. Vous pouvez signaler les effets secondaires à la FDA au 1-800-FDA-1088.
Informations sur les médicaments de la FDA
- Description de la drogue
- Indications
- Effets secondaires
- Avertissements
- Surdosage
- Pharmacologie clinique
- Guide des médicaments
Description de Vyondys 53
L'injection de Vyondys 53 (Golodirsen) est une solution concentrée sans conservation aqueuse stérile pour la dilution avant l'administration intraveineuse. Vyondys 53 est un liquide incolore clair à légèrement opalescent. Vyondys 53 est fourni dans des flacons à dose unique contenant 100 mg de golodirsen (50 mg / ml). Vyondys 53 est formulé comme une solution saline tamponnée au phosphate isotonique avec une osmolalité de 260 à 320 mOSM et un pH de 7,5. Chaque millilitre de Vyondys 53 contient: 50 mg de golodirsn; 0,2 mg de chlorure de potassium; 0,2 mg monobasique phosphate de potassium; 8 mg de chlorure de sodium; et 1,14 mg de phosphate de sodium dibasique anhydre dans l'eau pour l'injection. Le produit peut contenir de l'acide chlorhydrique ou de l'hydroxyde de sodium pour ajuster le pH.
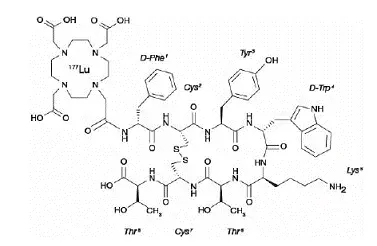
Le Golodirsen est un oligonucléotide antisens de la sous-classe phosphorodiamidate de morpholino (PMO). Les PMO sont des molécules synthétiques dans lesquelles les anneaux de ribofuranosyl à cinq membres trouvés dans l'ADN naturel et l'ARN sont remplacés par un cycle morpholino à six chaînons. Chaque cycle morpholino est lié par une fraction phosphorodiamidate non chargée plutôt que par la liaison phosphate chargée négativement qui est présente dans l'ADN nature et l'ARN. Chaque sous-unité morpholino phosphorodiamidate contient l'une des bases hétérocycliques trouvées dans l'ADN (adénine cytosine guanine ou thymine). Golodirsen contient 25 sous-unités liées. La séquence de bases de l'extrémité de 5 'à 3' est gttgcctcgggttctgaaggtgttc. La formule moléculaire de Golodirsen est C305H481N138O112P25 et le poids moléculaire est de 8647,28 daltons. La structure de Golodirsen est:
 |
Utilisations pour Vyondys 53
Vyondys 53 est indiqué pour le traitement de Dystrophie musculaire de Duchenne (DMD) chez les patients qui ont une mutation confirmée du gène DMD qui se prête à l'exon 53 saut. This indication is approved under accelerated approval based on an increase in dystrophin production in skeletal muscle observed in patients treated with Vyondys 53 [see Études cliniques ]. L'approbation continue de cette indication peut être subordonnée à la vérification d'un avantage clinique dans les essais de confirmation.
Dosage pour vyondys 53
Surveillance pour évaluer la sécurité
La jauge d'urine de la cystatine sérique et le rapport protéine / protéine urinaire-créatinine doivent être mesurées avant de démarrer Vyondys 53. Envisagez la mesure du taux de filtration glomérulaire avant le début de Vyondys 53. La surveillance de la toxicité rénale pendant le traitement est recommandée. Obtenez les échantillons d'urine avant la perfusion de Vyondys 53 ou au moins 48 heures après la perfusion la plus récente [voir Avertissements et précautions ].
Dossing Information
La dose recommandée de Vyondys 53 est de 30 milligrammes par kilogramme administré une fois par semaine en perfusion intraveineuse de 35 à 60 minutes via un filtre à 0,2 micron en ligne.
Si une dose de Vyondys 53 est manquée, elle peut être administrée dès que possible après la dose prévue.
Instructions de préparation
Vyondys 53 est fourni dans des flacons à dose unique comme une solution concentrée sans conservateur qui nécessite une dilution avant l'administration. Les médicaments parentéraux doivent être inspectés visuellement pour les particules et la décoloration avant l'administration chaque fois que la solution et le permis de conteneur. Utilisez la technique aseptique.
- Calculez la dose totale de Vyondys 53 à administrer en fonction du poids du patient et de la dose recommandée de 30 milligrammes par kilogramme. Déterminez le volume de Vyondys 53 nécessaire et le nombre correct de flacons pour fournir la dose calculée complète.
- Laissez les flacons se réchauffer à température ambiante. Mélanger le contenu de chaque flacon en inversant doucement 2 ou 3 fois. Ne secouez pas.
- Inspectez visuellement chaque flacon de Vyondys 53. La solution est un liquide incolore clair à légèrement opalescent et peut contenir des traces de petites particules amorphes blanches à blanches. N'utilisez pas si la solution dans les flacons est décollée nuageuse ou contient des particules étrangères autres que des traces de petites particules amorphes blanches à blanches.
- Avec une seringue équipée d'une aiguille de calibre 21 ou plus petite, retirez le volume calculé de Vyondys 53 du nombre approprié de flacons.
- Diluer le Vyondys retiré 53 dans l'injection de chlorure de sodium à 0,9% pour faire un volume total de 100 à 150 ml. Inversez doucement 2 à 3 fois pour mélanger. Ne secouez pas. Inspectez visuellement la solution diluée. N'utilisez pas si la solution est décollée nuageuse ou contient des particules étrangères autres que des traces de petites particules amorphes blanches à blanches.
- Administrer la solution diluée via un filtre à 0,2 micron en ligne.
- Vyondys 53 ne contient aucun conservateur et doit être administré immédiatement après la dilution. Perfusion complète de vyondys dilués 53 dans les 4 heures suivant la dilution. Si une utilisation immédiate n'est pas possible, le produit dilué peut être stocké jusqu'à 24 heures à 2 ° C à 8 ° C (36 ° F à 46 ° F). Ne congelez pas. Jeter les vyondys inutilisés 53.
Instructions d'administration
L'application d'une crème anesthésique topique au site de perfusion avant l'administration de Vyondys 53 peut être prise en compte.
Vyondys 53 est administré par perfusion intraveineuse. Flush la ligne d'accès intraveineuse avec 0,9% d'injection de chlorure de sodium USP avant et après la perfusion.
Infuser les Vyondys dilués 53 sur 35 à 60 minutes via un filtre à 0,2 micron en ligne. Ne mélangez pas d'autres médicaments avec Vyondys 53 ou infusez les autres médicaments par concomitance via la même ligne d'accès intraveineuse avec Vyondys 53.
Si une réaction d'hypersensibilité se produit, envisagez de ralentir la perfusion interrompant ou interrompant la thérapie Vyondys 53 [voir Contre-indications Avertissements et précautions et Effets indésirables ].
Comment fourni
Formes et forces posologiques
Vyondys 53 est un liquide incolore clair à légèrement opalescent et peut contenir des traces de petites particules amorphes blanches à blanches et disponibles en tant que:
- Injection : 100 mg / 2 ml (50 mg / ml)
Vyondys 53 L'injection est fournie dans des flacons à dose unique. La solution est un liquide incolore clair à légèrement opalescent et peut contenir des traces de petites particules amorphes blanches à blanches.
Flacons à dose unique contenant 100 mg / 2 ml (50 mg / ml) NDC 60923-465-02
Stockage et manipulation
Stockez les vyondys 53 à 2 ° C à 8 ° C (36 ° F à 46 ° F). Ne congelez pas. Stockez en carton d'origine jusqu'à ce qu'il soit prêt à utiliser pour protéger de la lumière.
Fabriqué pour: Sarepta Therapeutics Inc. Cambridge MA 02142 USA. Révisé: juin 2024
Effets secondaires pour Vyondys 53
Les réactions indésirables graves suivantes sont décrites ci-dessous et ailleurs dans l'étiquetage:
- Réactions d'hypersensibilité [voir Avertissements et précautions ]
Expérience des essais cliniques
Étant donné que les essais cliniques sont menés dans des conditions de réaction indésirables très variables observées dans les essais cliniques d'un médicament ne peuvent pas être directement comparées aux taux dans les essais cliniques d'un autre médicament et ne peuvent pas refléter les taux observés dans la pratique.
Dans le programme de développement clinique de Vyondys 53 58 patients ont reçu au moins une dose intraveineuse de Vyondys 53 allant entre 4 mg / kg (0,13 fois le dosage recommandé) et 30 mg / kg (le dosage recommandé). Tous les patients étaient des hommes et avaient une dystrophie musculaire de Duchenne génétiquement confirmée. L'âge à l'entrée de l'étude était de 6 à 13 ans. La plupart (86%) patients étaient du race blanche.
Vyondys 53 was studied in 2 double-blind placebo-controlled studies.
Dans l'étude 1, la partie 1, les patients ont été randomisées pour recevoir des perfusions intraveineuses une fois par semaine de Vyondys 53 (n = 8) dans quatre niveaux de dose croissants de 4 mg / kg à 30 mg / kg ou placebo (n = 4) pendant au moins 2 semaines à chaque niveau. Tous les patients qui ont participé à l'étude 1 partie 1 (n = 12) ont été poursuivis dans l'étude 1 partie 2 Une extension ouverte au cours desquelles ils ont reçu Vyondys 53 à une dose de 30 mg / kg IV une fois par semaine [voir Études cliniques ].
Dans l'étude, 2 patients ont reçu des Vyondys 53 (n = 33) 30 mg / kg ou un placebo (n = 17) iv une fois par semaine jusqu'à 96 semaines après quoi tous les patients ont reçu des Vyondys 53 à une dose de 30 mg / kg.
Les effets indésirables observés chez au moins 20% des patients traités dans les sections contrôlées par placebo des études 1 et 2 sont présentées dans le tableau 1.
Tableau 1: Réactions indésirables qui se sont produites chez au moins 20% des patients traités à 53 Vyondys et à un rythme supérieur au placebo dans les études 1 et 2
| Réaction indésirable | Vyondys 53 (N = 41)% | Placebo (N = 21)% |
| Mal de tête | 41 | 10 |
| Pyrexie | 41 | 14 |
| Automne | 29 | 19 |
| Douleurs abdominales | 27 | 10 |
| Nasopharynngite | 27 | 14 |
| Toux | 27 | 19 |
| Vomissement | 27 | 19 |
| Nausée | 20 | 10 |
Autres réactions indésirables qui se sont produites à une fréquence supérieure à 5% des patients traités à 53 Vyondy allergie Tachycardie Cathéter Site Reaction Constipation and Fracture.
Des réactions d'hypersensibilité se sont produites chez les patients traités avec Vyondys 53 [voir Avertissements et précautions ].
Quelle est la quantité de DXM dans Robitussin
Expérience de commercialisation de la poste
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation de Vyondys 53. Parce que ces réactions sont signalées volontairement à partir d'une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale à l'exposition au médicament.
Troubles du système immunitaire: anaphylaxie [voir Contre-indications et Avertissements et précautions ]
Interactions médicamenteuses pour Vyondys 53
Aucune information fournie
Avertissements pour Vyondys 53
Inclus dans le cadre du PRÉCAUTIONS section.
Précautions pour Vyondys 53
Réactions d'hypersensibilité
Des réactions d'hypersensibilité, notamment l'anaphylaxie, la dermatite de l'irticaire de la pyrexie de l'irticaire et l'exfoliation cutanée se sont produites chez les patients traités par Vyondys 53 nécessitant un traitement. Si une réaction d'hypersensibilité se produit instituer un traitement médical approprié et envisager de ralentir l'interruption ou d'arrêter la thérapie Vyondys 53 et de surveiller jusqu'à ce que la condition résout [voir Posologie et administration ]. Vyondys 53 is contraindicated in patients with a history of a serious hypersensitivity reaction to golodirsen or to any of the inactive ingredients in Vyondys 53 [see Contre-indications ].
Toxicité rénale
Une toxicité rénale a été observée chez les animaux qui ont reçu Golodirsen [voir Utiliser dans des populations spécifiques ]. Although kidney toxicity was not observed in the clinical studies with Vyondys 53 the clinical experience with Vyondys 53 is limited et kidney toxicity including potentially fatal glomerulonephritis has been observed after administration of some antisense oligonucleotides. Kidney function should be monitored in patients taking Vyondys 53. Because of the effect of reduced skeletal muscle mass on creatinine measurements creatinine may not be a reliable measure of kidney function in DMD patients. Serum cystatin C urine dipstick et urine protein-to-creatinine ratio should be measured before starting Vyondys 53. Consider also measuring glomerular filtration rate using an exogenous filtration marker before starting Vyondys 53. During treatment monitor urine dipstick every month et serum cystatin C et urine protein-to-creatinine ratio every three months. Only urine expected to be free of excreted Vyondys 53 should be used for monitoring of urine protein. Urine obtained on the day of Vyondys 53 infusion prior to the infusion or urine obtained at least 48 hours after the most recent infusion may be used. Alternatively use a laboratory test that does not use the reagent pyrogallol red as this reagent has the potential to cross react with any Vyondys 53 that is excreted in the urine et thus lead to a false positive result for urine protein.
Si une augmentation persistante de la cystatine C ou de la protéinurie sérique est détectée, reportez-vous à un néphrologue pédiatrique pour une évaluation plus approfondie.
Toxicologie non clinique
Carcinogenèse Mutagenèse A trouble de la fertilité
Cancérogenèse
L'administration de souris Golodirsen en souris transgéniques mâles (Tg.Rash2) (0 100 300 ou 1000 mg / kg) par semaine pendant 26 semaines par injection sous-cutanée et à des rats mâles (0 30 100 ou 300 mg / kg) chaque semaine jusqu'à 102 semaines par injection intraveineuse n'a entraîné aucune augmentation des néoplasmes.
Mutagenèse
Le golodirsen était négatif dans les tests in vitro (mutation bactérienne inverse et aberration chromosomique dans les cellules CHO) et in vivo (micronucleus de moelle osseuse de souris).
Altération de la fertilité
Les études de fertilité chez les animaux n'ont pas été menées avec Golodirsen. Aucun effet de Golodirsen sur le système de reproduction masculin n'a été observé après l'administration sous-cutanée hebdomadaire (0 120 300 ou 600 mg / kg) à des souris mâles ou à une administration intraveineuse hebdomadaire (0 80 200 ou 400 mg / kg) à des singes mâles. dose intraveineuse de 30 mg / kg.
Utiliser dans des populations spécifiques
Grossesse
Résumé des risques
Il n'y a pas de données humaines ou animales disponibles pour évaluer l'utilisation de Vyondys 53 pendant la grossesse. Aux États-Unis, les principales malformations congénitales de la population générale se produisent dans 2 à 4% et une fausse couche se produit dans 15 à 20% des grossesses cliniquement reconnues.
Lactation
Résumé des risques
Il n'y a pas de données humaines ou animales pour évaluer l'effet de Vyondys 53 sur la production de lait la présence de Golodirsen dans le lait ou les effets de Vyondys 53 sur le nourrisson allaité.
Les avantages du développement et de la santé de l'allaitement doivent être pris en compte avec le besoin clinique de la mère pour Vyondys 53 et tout effet indésirable potentiel sur le nourrisson allaité de Vyondys 53 ou de l'état maternel sous-jacent.
Usage pédiatrique
Vyondys 53 est indiqué pour le traitement de Dystrophie musculaire de Duchenne (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping including pediatric patients [see Études cliniques ].
L'administration intraveineuse de golodirsen (0 100 300 ou 900 mg / kg) aux rats mâles juvéniles une fois par semaine pendant 10 semaines (jours postnatals 14 à 77) n'a pas entraîné de toxicité postnatale de développement (par exemple la fonction immunitaire neurobehaviorale ou de reproduction masculine). Cependant, à la dose la plus élevée testée (900 mg / kg / semaine), le Golodirsen a entraîné la mort d'animaux en raison d'une insuffisance rénale ou d'une défaillance. Chez les animaux survivants (dont un animal à la dose la plus faible testée), il y a eu une augmentation de la dose-dépendante de l'incidence et de la gravité des effets tubulaires rénaux (y compris la dégénérescence / régénération de la fibrose et la dilatation) qui étaient corrélées avec les changements dans les paramètres de pathologie clinique reflétant une altération dose-dépendante de la fonction rénale. De plus, une diminution de la teneur en minéraux et de la densité minérale de la zone osseuse a été observée à la dose la plus élevée testée (900 mg / kg semaine) mais sans effet sur la croissance osseuse. Une dose sans effet pour la toxicité rénale n'a pas été identifiée; La dose la plus faible testée (100 mg / kg / semaine) a été associée à des expositions au plasma (ASC) environ 2,5 fois chez l'homme à la dose humaine recommandée de 30 mg / kg / semaine.
Utilisation gériatrique
Le DMD est en grande partie une maladie des enfants et des jeunes adultes; Par conséquent, il n'y a pas d'expérience gériatrique avec Vyondys 53.
Patients souffrant de troubles rénaux
La clairance rénale du golodirsen est réduite chez les adultes non DMD présentant une déficience rénale basée sur le taux de filtration glomérulaire estimé calculé en utilisant la modification de l'équation de l'alimentation et des maladies rénales (MDRD) [voir Pharmacologie clinique ]. However because of the effect of reduced skeletal muscle mass on creatinine measurements in DMD patients no specific dosage adjustment can be recommended for DMD patients with renal impairment based on estimated glomerular filtration rate. Patients with known renal function impairment should be closely monitored during treatment with Vyondys 53.
Informations sur la surdose pour Vyondys 53
Aucune information fournie
Contre-indications pour Vyondys 53
Vyondys 53 is contraindicated in patients with a serious hypersensitivity reaction to golodirsen or to any of the inactive ingredients in Vyondys 53. Anaphylaxis has occurred in patients receiving Vyondys 53 [see Avertissements et précautions ].
Pharmacologie clinique for Vyondys 53
Mécanisme d'action
Golodirsen est conçu pour se lier à l'exon 53 de la dystrophine pré-ARNm, ce qui entraîne l'exclusion de cet exon pendant le traitement de l'ARNm chez les patients présentant des mutations génétiques qui se prêtent à l'exon 53 saut. Le saut d'exon 53 est destiné à permettre la production d'une protéine de dystrophine tronquée en interne chez les patients atteints de mutations génétiques qui se prêtent à l'exon 53 saut [voir Études cliniques ].
Pharmacodynamique
Après traitement avec Vyondys 53 Tous les patients ont évalué (n = 25) dans l'étude 1 partie 2 [voir Études cliniques ] a eu une augmentation de la sauté de l'exon 53 démontré par la réaction en chaîne de polymérase de transcription inverse (RT-PCR) par rapport à la ligne de base.
Dans l'étude 1 partie 2 [voir Études cliniques ] Les niveaux de dystrophine évalués par le test Sarepta Western blot sont passés de 0,10% (ET 0,07) de normale à la ligne de base à 1,02% (ET 1,03) de la normale après 48 semaines de traitement avec Vyondys 53. Le changement moyen par rapport à la dystrophine après 48 semaines de traitement (P.<0.001); the median change from baseline was 0.88%. This increase in dystrophin protein expression positively correlated with the level of exon skipping. Dystrophin levels assessed by western blot can be meaningfully influenced by differences in sample processing analytical technique reference materials et quantitation methodologies. Therefore comparing dystrophin results from different assay protocols will require a stetardized reference material et additional bridging studies.
La localisation correcte de la dystrophine tronquée au sarcolemme dans les fibres musculaires des patients traitées par golodirsen a été démontrée par coloration par immunofluorescence.
Pharmacocinétique
La pharmacocinétique de Golodirsen a été évaluée chez les patients DMD après l'administration de doses intraveineuses allant de 4 mg / kg / semaine à 30 mg / kg / semaine (c'est-à-dire la posologie recommandée). L'exposition à Golodirsen a augmenté proportionnellement à la dose avec une accumulation minimale avec un dosage une fois par semaine. La variabilité inter-sujet (en% CV) pour CMAX et AUC variait de 38% à 72% et 34% à 44% respectivement.
Distribution
Le volume de distribution à l'état d'équilibre était similaire entre les patients DMD et les sujets sains. Le volume de distribution à l'état d'équilibre de Golodirsen moyen était de 668 ml / kg (% CV = 32,3) à une dose de 30 mg / kg. La liaison aux protéines plasmatiques de Golodirsen variait de 33% à 39% et ne dépend pas de la concentration.
Élimination
La demi-vie d'élimination de Golodirsen (SD) était de 3,4 (0,6) heures et le dégagement du plasma était de 346 ml / h / kg à la dose de 30 mg / kg.
Métabolisme
Golodirsen est métaboliquement stable. Aucun métabolite n'a été détecté dans le plasma ou l'urine.
Excrétion
Golodirsen est principalement excrété inchangé dans l'urine. La demi-vie d'élimination (T½) était de 3,4 heures.
Populations spécifiques
Âge
La pharmacocinétique de Golodirsen a été évaluée chez des patients atteints de DMD pédiatriques masculins. Il n'y a aucune expérience de l'utilisation de Vyondys 53 chez les patients DMD 65 ans ou plus.
Sexe
Sexe effects have not been evaluated; Vyondys 53 has not been studied in female patients.
Course
L'impact potentiel de la race n'est pas connu car 92% des patients en études étaient des Caucasiens.
Patients souffrant de troubles rénaux
L'effet de la déficience rénale sur la pharmacocinétique de Golodirsen a été évalué chez des sujets non DMD âgés de 41 à 65 ans avec une maladie rénale chronique de stade 2 (CKD) (n = 8 taux de filtration glomérulaire estimé (EGFR) ≥60 et <90 mL/min/1.73 m²) or Stage 3 CKD (n=8 eGFR ≥30 et <60 mL/min/1.73 m²) et matched healthy subjects (n=8 eGFR ≥90 mL/min/1.73 m²). Subjects received a single 30 mg/kg IV dose of golodirsen.
Chez les sujets présentant une exposition aux CKD de stade 2 ou de stade 3 (ASC) ont augmenté respectivement de 1,2 fois et 1,9 fois. Il n'y a eu aucun changement dans le CMAX chez les sujets avec des CKD de stade 2; Chez les sujets atteints de CKD de stade 3, il y a eu une augmentation de 1,2 fois de la CMAX par rapport aux sujets avec une fonction rénale normale. L'effet de l'étape 4 ou de l'étape 5 des CKD sur la pharmacocinétique et la sécurité de Golodirsen n'a pas été étudié.
Les valeurs de DMF estimées dérivées des équations de MDRD et les définitions de seuil de divers stades de CKD chez des adultes par ailleurs en bonne santé ne seraient pas généralisables aux patients pédiatriques atteints de DMD. Par conséquent, aucun ajustement posologique spécifique ne peut être recommandé pour les patients souffrant de troubles rénaux [voir Utiliser dans des populations spécifiques ].
Patients souffrant de troubles hépatiques:
Vyondys 53 has not been studied in patients with hepatic impairment.
Études d'interaction médicamenteuse
Golodirsen n'a pas inhibé CYP1A2 CYP2B6 CYP2C8 CYP2C9 CYP2C19 CYP2D6 ou CYP3A4 / 5 in vitro. Le Golodirsen était un faible inducteur de CYP1A2 et n'a pas induit CYP2B6 ou CYP3A4. Le Golodirsen n'a pas été métabolisé par les microsomes hépatiques humains et n'était pas un substrat ou un fort inhibiteur de l'un des principaux transporteurs de médicaments humains testés (OAT1 OAT3 OCT2 OATP1B1 MATE1 P-GP BCRP et MRP2 OATP1B3 et MATE2-K). Sur la base des données in vitro, le Golodirsen a un faible potentiel d'interactions médicamenteuses chez l'homme.
Toxicologie et / ou pharmacologie animale
Une toxicité rénale a été observée dans des études chez des souris mâles et des rats; Des résultats de la vessie urinaire ont été observés chez les souris mâles.
Chez les souris mâles, Golodisen a été administré chaque semaine pendant 12 semaines par injection intraveineuse (0 12120 ou 960 mg / kg) ou pendant 26 semaines par injection sous-cutanée (0 120 300 ou 600 mg / kg). Dans l'étude de 12 semaines, les résultats microscopiques dans les reins (dilatation tubulaire basophile ou la vacuolation des coulées éosinophiles) étaient corrélées à l'augmentation des marqueurs sériques de la fonction rénale (par ex. urée L'azote créatinine) ont été observés principalement à la dose la plus élevée testée; L'hypertrophie de l'épithélium transitionnel de l'uretère ou de la vessie a été observée à toutes les doses. Dans l'étude de 26 semaines, la dégénérescence tubulaire rénale et la dégénérescence de l'épithélium transitionnel de la vessie urinaire ont été observées à toutes les doses.
Chez les rats mâles, l'administration intraveineuse de Golodirsen (0 60 100 300 ou 600 mg / kg) par semaine pendant 13 semaines a entraîné une dégénérescence tubulaire, sauf la dose la plus faible testée; À la dose élevée, les changements microscopiques se sont accompagnés d'une augmentation de l'azote de l'urée sérique.
Chez les singes masculins, l'administration intraveineuse de Golodirsen (0 80 200 ou 400 mg / kg) par semaine pendant 39 semaines a entraîné des changements microscopiques dans la dilatation rénale (basophilie ou infiltration des cellules mononucléaires) à toutes les doses qui corrélaient avec l'augmentation des marqueurs de sérum de la fonction du rénal (créatinine d'urée nitrogén) à l'atteinte du plus haut dose.
Études cliniques
L'effet de Vyondys 53 sur la production de dystrophine a été évalué dans une étude chez les patients DMD avec une mutation confirmée du gène DMD qui se prête à l'exon 53 saut (étude 1; NCT02310906).
L'étude 1 partie 1 était une étude de dose-titration contrôlée par placebo en double aveugle chez 12 patients DMD. Les patients ont été randomisés 2: 1 pour recevoir Vyondys 53 ou un placebo correspondant. Les patients Vyondys 53âts ont reçu quatre niveaux de dose croissants allant de 4 mg / kg / semaine (moins que le dosage recommandé) à 30 mg / kg / semaine par perfusion intraveineuse pendant 2 semaines à chaque niveau de dose.
Combien de soma est trop
L'étude 1 partie 2 était une étude ouverte de 168 semaines évaluant l'efficacité et l'innocuité de Vyondys 53 à une dose de 30 mg / kg / semaine chez les 12 patients inscrits dans la partie 1 plus 13 patients naïfs supplémentaires avec DMD, étant entièrement exoné par l'exon 53. À l'entrée de l'étude (dans la partie 1 ou la partie 2), les patients ont eu un âge médian de 8 ans et étaient sur une dose stable de corticostéroïdes pendant au moins 6 mois. L'efficacité a été évaluée en fonction du changement par rapport à la ligne de base dans le niveau de protéine de dystrophine (mesurée en% du niveau de dystrophine chez des sujets sains, c'est-à-dire un% de la normale) à la semaine 48 de la partie 2. Les biopsies musculaires ont été obtenues au départ avant le traitement et à la semaine 48 de la partie 2 de tous les niveaux de la Bot Western (n = 25) de la semaine 48 de la BLOT. Les niveaux moyens de dystrophine sont passés de 0,10% (ET 0,07) de la normale au départ à 1,02% (ET 1,03) de la normale à la semaine 48 de l'étude 1 partie 2 avec un changement moyen de la dystrophine de 0,92% (SD 1,01) des niveaux normaux (P (P<0.001); the median change from baseline was 0.88%.
Les niveaux de dystrophine individuelle des patients de l'étude 1 sont présentés dans le tableau 2.
Tableau 2: Expression de la dystrophine Sarepta Western blot par le patient individuel de l'étude 1
| Numéro de patient | Sarepta Western blot% dystrophine normale | Numéro de patient | Sarepta Western blot% dystrophine normale | ||||
| Base de base | Partie 2 semaine 48 | Changement de la ligne de base | Base de base | Partie 2 semaine 48 | Changement de la ligne de base | ||
| 1 | 0.08 | 0.09 | 0.01 | 14 | 0.22 | 0.28 | 0.06 |
| 2 | 0.11 | 0.11 | 0.01 | 15 | 0.14 | 0.21 | 0.07 |
| 3 | 0.21 | 0.22 | 0.01 | 16 | 0.05 | 0.42 | 0.37 |
| 4 | 0.05 | 0.12 | 0.08 | 17 | 0.07 | 1.03 | 0.97 |
| 5 | 0.03 | 0.12 | 0.09 | 18 | 0.02 | 1.57 | 1.55 |
| 6 | 0.06 | 0.14 | 0.09 | 19 | 0.12 | 1.17 | 1.05 |
| 7 | 0.12 | 0.37 | 0.25 | 20 | 0.03 | 1.72 | 1.69 |
| 8 | 0.11 | 1.06 | 0.95 | 21 | 0.11 | 1.77 | 1.66 |
| 9 | 0.06 | 0.54 | 0.48 | 22 | 0.31 | 4.30 | 3.99 |
| 10 | 0.05 | 0.97 | 0.92 | 23 | 0.11 | 0.36 | 0.25 |
| 11 | 0.06 | 1.55 | 1.49 | 24 | 0.03 | 0.91 | 0.88 |
| 12 | 0.07 | 1.91 | 1.84 | 25 | 0.07 | 1.29 | 1.22 |
| 13 | 0.10 | 3.25 | 3.15 |
Informations sur les patients pour Vyondys 53
Réactions d'hypersensibilité
Conseiller les patients et / ou les soignants que les réactions d'hypersensibilité, notamment la dermatite et l'exfoliation cutanée de la pyrexie de l'irticaire et l'exfoliation cutanée, les patients ont été traités avec des vyondys 53. Avertissements et précautions ].
Toxicité rénale
Informer les patients que la néphrotoxicité s'est produite avec des médicaments similaires à Vyondys 53. Conseillez les patients de l'importance de la surveillance de la toxicité rénale par leurs prestataires de soins de santé pendant le traitement avec Vyondys 53 [voir Avertissements et précautions ].