Lutathera
Les Informations Présentées Sur Ce Site Ne Constituent Pas Un Avis Médical. Nous Ne Vendons Rien. L'Exactitude De La Traduction N'Est Pas Garantie. Clause De Non-Responsabilité
Résumé
Qu'est-ce que Lutathera?
L'injection de Lutathera (lutetium Lu 177 dotatate) est un analogue de somatostatine radiomarqué indiqué pour le traitement des récepteurs de la somatostatine positifs gastro-entéropancréatiques tumeurs neuroendocrines (NI-NETS), y compris les tumeurs intermédiaires foregut et les tumeurs neuroendocrines hindous chez l'adulte.
Quels sont les effets secondaires de Lutathera?
Les effets secondaires courants de Lutathera comprennent:
- faible niveau de lymphocytes dans le sang (lymphopénie)
- Augmentation du GGT
- vomissement
- nausée
- augmentation de l'AST
- Augmentation de l'ALT
- glycémie ( hyperglycémie )
- bouscule potassium ( hypokaliémie )
- fatigue
- douleurs abdominales
- diarrhée
- diminution de l'appétit
- mal de tête
- vertiges
- gonflement ou douleur dans les extrémités
- bouffée
- maux de dos
- anxiété
- insuffisance rénale
- perte
- hypertension artérielle ( hypertension )
- tousser et
- constipation
Cherchez des soins médicaux ou appelez le 911 à la fois si vous avez les effets secondaires graves suivants:
- Des symptômes oculaires graves tels que la perte de vision soudaine floue du tunnel de vision de la vision de la vision des yeux ou de l'enflure ou de voir des halos autour des lumières;
- Symptômes cardiaques graves tels que les battements cardiaques rapides ou battants; flotter dans votre poitrine; essoufflement; et des étourdissements soudains étourdisseurs ou s'évanouissant;
- Maux de tête sévères Confusion Slurred Speech Bras ou Ligne Proulitude Trouble de la merde Perte de coordination Sentiment des muscles très rigides très rigides Fièvre élevée Propice ou des tremblements.
Ce document ne contient pas tous les effets secondaires possibles et d'autres peuvent survenir. Vérifiez auprès de votre médecin des informations supplémentaires sur les effets secondaires.
Dosage pour Lutathera
La dose de Lutathera est de 7,4 GBQ (200 MCI) administrée toutes les 8 semaines pour un total de 4 doses.
Quelles substances ou suppléments de médicaments interagissent avec Lutathera?
Lutathera peut interagir avec les analogues de la somatostatine. Dites à votre médecin tous les médicaments et suppléments que vous utilisez.
Lutathera pendant la grossesse et l'allaitement
Dites à votre médecin si vous êtes enceinte ou prévoyez de tomber enceinte avant d'utiliser Lutathera; Cela peut nuire à un fœtus. En raison du risque potentiel de réactions indésirables graves chez l'allaitement maternel allaité, il n'est pas recommandé d'utiliser Lutathera et pendant 2,5 mois après la dose finale.
Informations Complémentaires
Notre injection de Lutathera (lutetium Lu 177 dotatate) pour un centre de médicaments à effet secondaire à usage intraveineux offre une vue complète des informations sur les médicaments disponibles sur les effets secondaires potentiels lors de la prise de ce médicament.
Informations sur les médicaments de la FDA
- Description de la drogue
- Indications
- Effets secondaires
- Avertissements
- Surdosage
- Pharmacologie clinique
- Guide des médicaments
Description de Lutathera
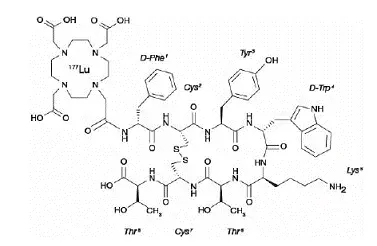
Lutathera (Lutetium Lu 177 dotatate) est un analogue de somatostatine radiomarqué. Le médicament substance lutetium Lu 177 dotatate est un peptide cyclique lié au chélateur lié de manière covalente 14710-tétraazacyclododécane-14710-tétraacétique à un radionucléide.
Le lutétium Lu 177 dotatate est décrit comme le lutetium (Lu 177) -N - [((4710-tricarboxyméthyl-14710-tétraazacyclododec (2-7) disulfure. Le poids moléculaire est de 1609,6 daltons et la formule structurelle est la suivante:
 |
Lutathera (lutetium Lu 177 dotatate) 370 MBQ / ml (10 mc / ml) est une solution stérile sans molle claire à légèrement jaune pour une utilisation par voie intraveineuse. Chaque flacon à dose unique contient de l'acide acétique (0,48 mg / ml) d'acétate de sodium (0,66 mg / ml) d'acide gentille (0,63 mg / ml) d'hydroxyde de sodium (0,65 mg / ml) d'acide ascorbique (2,8 mg / ml) de triamine de diéthylène (0,05 mg / ml) de sodium (6,85 pentaacétique (0,05 mg / ml) de sodium (6,85 mg / ml) et eau pour l'injection (AD 1 ml). La plage de pH de la solution est de 4,5 à 6.
Caractéristiques physiques
Le lutétium (Lu 177) se désintègre à un hafnium stable (HF 177) avec une demi-vie de 6,647 jours en émettant un rayonnement bêta avec une énergie maximale de 0,498 MeV et un rayonnement photonique (γ) de 0,208 MEV (11%) et 0,113 MEV (6,4%). Les radiations principales sont détaillées dans le tableau 6.
Tableau 6: Lu 177 Radiations principales
| Radiation | Énergie (kev) | IP% | Ig% |
| β- | 176.5 | 12.2 | |
| β- | 248.1 | 0.05 | |
| β- | 384.9 | 9.1 | |
| β- | 497.8 | 78.6 | |
| γ | 71.6 | 0.15 | |
| γ | 112.9 | 6.40 | |
| γ | 136.7 | 0.05 | |
| γ | 208.4 | 11.0 | |
| γ | 249.7 | 0.21 | |
| γ | 321.3 | 0.22 |
Rayonnement externe
Le tableau 7 résume les propriétés de désintégration radioactive de LU 177.
Tableau 7: Tableau de désintégration physique: Lutetium LU 177 demi-vie = 6,647 jours
| Heures | Fraction restante | Heures | Fraction restante |
| 0 | 1.000 | 48 (2 jours) | 0.812 |
| 1 | 0.996 | 72 (3 jours) | 0.731 |
| 2 | 0.991 | 168 (7 jours) | 0.482 |
| 5 | 0.979 | 336 (14 jours) | 0.232 |
| 10 | 0.958 | 720 (30 jours) | 0.044 |
| 24 (1 jour) | 0.901 | 1080 (45 jours) | 0.009 |
Utilisations pour Lutathera
Lutathera est indiquée pour le traitement des patients adultes et pédiatriques de 12 ans et plus avec des tumeurs neuroendocrines gastroentéropancréatiques (GEP-NET), des tumeurs de neuroendocrine intermédiaire et de neuroendocrine Hindoue.
Dosage pour Lutathera
Instructions de sécurité importantes
Lutathera est un radiopharmaceutique; manipuler avec des mesures de sécurité appropriées pour minimiser l'exposition aux rayonnements [voir Avertissements et précautions ]. Utilisez des gants imperméables et un blindage de rayonnement efficace lors de la manipulation de Lutathera. Les radiopharmaceutiques, y compris Lutathera, doivent être utilisés par ou sous le contrôle des prestataires de soins de santé qui sont qualifiés par une formation et une expérience spécifiques dans l'utilisation et la manipulation sûres des radiopharmaceutiques et dont l'expérience et la formation ont été approuvées par l'agence gouvernementale appropriée autorisée à concéder à l'utilisation des radiopharmaceutiques.
Vérifiez le statut de grossesse des femmes de potentiel reproducteur avant de lancer Lutathera [voir Utiliser dans des populations spécifiques ].
Surveillez de près les patients pour les signes et symptômes de réactions d'hypersensibilité pendant et après l'administration de Lutathera pendant au moins 2 heures dans un cadre où des médicaments et équipements de réanimation cardiopulmonaire sont disponibles [voir Avertissements et précautions ].
Dosage recommandé
La dose de Lutathera recommandée pour les patients adultes et pédiatriques de 12 ans et plus est de 7,4 GBQ (200 MCI) toutes les 8 semaines (± 1 semaine) pour un total de 4 doses. Administrer des prémédications et des médicaments concomitants comme recommandé [voir Prémédications et médicaments concomitants ].
Prémédications et médicaments concomitantsAnalogues de somatostatine
- Avant de lancer un traitement de Lutathera: interrompre les analogues de la somatostatine à action prolongée (par exemple l'octréotide à action prolongée) au moins 4 semaines avant de lancer Lutathera. Administrer l'octréotide à courte durée d'action au besoin; abandonner au moins 24 heures avant de lancer Lutathera [voir Interactions médicamenteuses ].
- Pendant le traitement de Lutathera: administrer l'octréotide à action longue 30 mg par voie intramusculaire entre 4 et 24 heures après chaque dose de Lutathera. N'administrez pas l'octréotide à longue durée d'action dans les 4 semaines précédant chaque dose de Lutathera ultérieure. L'octréotide à courte durée d'action peut être donné pour la gestion des symptômes pendant le traitement de Lutathera, mais doit être retenu au moins 24 heures avant chaque dose de Lutathera.
- Après le traitement de Lutathera: Continuez l'octréotide à action prolongée 30 mg par voie intramusculaire toutes les 4 semaines après avoir terminé Lutathera jusqu'à la progression de la maladie ou pendant 18 mois après l'initiation du traitement à la discrétion du médecin.
Antiémétiques
Administrer des antiémétiques avant la solution d'acides aminés recommandée.
Solution d'acide aminé
Initiez une perfusion intraveineuse d'une solution d'acides aminés stérile contenant de la L-lysine et de la L-arginine (tableau 1) 30 minutes avant le début de la perfusion de Lutathera. Utilisez une valve à trois voies pour administrer la solution d'acides aminés en utilisant le même accès veineux que Lutathera ou administrer la solution d'acide aminé par un accès veineux séparé dans l'autre bras du patient. Continuez la perfusion de la solution d'acide aminé pendant et pendant au moins 3 heures après la fin de la perfusion de Lutathera. Ne diminuez pas la dose de la solution d'acides aminés si une dose réduite de Lutathera est administrée [voir Avertissements et précautions ].
Tableau 1. Solution d'acide aminé
| Article | Spécification |
| L-lysine hcl | Entre 18 et 25 ga |
| L-arginine HCL | Entre 18 et 25 gb |
| Volume | 1 à 2 L |
| Osmolalité | <1200 mOsmol/kg |
| aÉquivalent à 14,4 à 20 g l-lysine. bÉquivalent à 14,9 à 20,7 g de L-arginine. |
Hypersensibilité prophylaxie
Les patients prémédicaux qui ont eu des réactions d'hypersensibilité antérieures de grade 1 ou 2 à Lutathera. Ne réprimandez pas les patients qui éprouvent des réactions d'hypersensibilité de grade 3 ou 4 à Lutathera [voir Avertissements et précautions ].
Modifications posologiques pour les effets indésirables
Les modifications de dose recommandées de Lutathera pour les effets indésirables sont fournies dans le tableau 2.
Tableau 2. Modifications de dosage recommandées de Lutathera pour Effets indésirables
| Réaction indésirable | a | Modification de la dose |
| Thrombocytopénie [voir Avertissements et précautions ] | Première occurrence de grade 2 3 ou 4 | Retenir la dose jusqu'à une résolution complète ou partielle (grade 0 à 1). Reprenez Lutathera à 3,7 GBQ (100 MCI) chez les patients présentant une résolution complète ou partielle. Si la dose réduite n'entraîne pas de grade 2 3 ou 4 thrombocytopénie, administrer Lutathera à 7,4 GBQ (200 MCI) comme dose suivante. Arrêter en permanence Lutathera pour la thrombocytopénie de grade 2 ou plus, nécessitant un intervalle de dosage au-delà de 16 semaines. |
| Recurrent Grade 2 3 ou 4 | Arrêter de façon permanente Lutathera. | |
| Anémie et neutropénie [voir Avertissements et précautions ] | Première occurrence de grade 3 ou 4 | Retenir la dose jusqu'à une résolution complète ou partielle (grade 0 1 ou 2). Reprenez Lutathera à 3,7 GBQ (100 MCI) chez les patients présentant une résolution complète ou partielle. Si une dose réduite n'entraîne pas une anémie ou 4 de grade ou 4, administrer Lutathera à 7,4 GBQ (200 MCI) comme dose suivante. Arrêter de façon permanente Lutathera pour une anémie ou une neutropénie supérieure ou plus, nécessitant un intervalle de dosage au-delà de 16 semaines. |
| Recurrent Grade 3 ou 4 | Arrêter de façon permanente Lutathera. | |
| Toxicité rénale [voir Avertissements et précautions ] | Première occurrence de:
| Retenir la dose jusqu'à la résolution ou revenir à la ligne de base. Reprenez Lutathera à 3,7 GBQ (100 MCI) chez les patients résolution ou retour à la ligne de base. Si une dose réduite n'entraîne pas la toxicité rénale, l'administration de Lutathera à 7,4 GBQ (200 MCI) comme dose suivante. Arrêter en permanence Lutathera pour la toxicité rénale nécessitant un intervalle de dosage au-delà de 16 semaines. |
| Toxicité rénale récurrente | Arrêter de façon permanente Lutathera. | |
| Hépatotoxicité [voir Avertissements et précautions ] | Première occurrence de:
| Retenir la dose jusqu'à la résolution ou revenir à la ligne de base. Reprenez Lutathera à 3,7 GBQ (100 MCI) chez les patients résolution ou retour à la ligne de base. En cas de réduction de la dose de Lutathera, n'entraîne pas l'hépatotoxicité administrer Lutathera à 7,4 GBQ (200 MCI) comme dose suivante. Arrêter en permanence Lutathera pour l'hépatotoxicité nécessitant un intervalle de dosage au-delà de 16 semaines. |
| Hépatotoxicité récurrente | Arrêter de façon permanente Lutathera. | |
| Réactions d'hypersensibilitéb[voir Avertissements et précautions ] | Première occurrence de grade 3 ou 4 | Arrêter de façon permanente Lutathera. |
| Toute autre réaction indésirablec [voir Effets indésirables ] | Première occurrence de grade 3 ou 4 | Retenir la dose jusqu'à une résolution complète ou partielle (grade 0 à 2). Reprenez Lutathera à 3,7 GBQ (100 MCI) chez les patients présentant une résolution complète ou partielle. Si la dose réduite n'entraîne pas la toxicité de 3 ou 4, administrez Lutathera à 7,4 GBQ (200 MCI) comme dose suivante. Arrêter en permanence Lutathera pour les effets indésirables de grade 3 ou plus nécessitant un intervalle de dosage au-delà de 16 semaines. |
| Recurrent Grade 3 ou 4 | Arrêter de façon permanente Lutathera. | |
| aLe classement de la gravité est défini dans les critères de terminologie courants les plus récents pour les événements indésirables (CTCAE). bY compris la réaction allergique et l'anaphylaxie. cAucune modification de la dose requise pour les toxicités hématologiques de grade 3 ou de grade 4 uniquement en raison de la lymphopénie. |
Préparation et administrationInstructions de préparation
- Utilisez la technique aseptique et le blindage des rayonnements lors de la manipulation ou de l'administration de la solution de Lutathera. Utilisez des pinces lors de la manipulation du flacon pour minimiser l'exposition aux rayonnements.
- Inspectez le produit visuellement sous un écran blindé pour les particules et la décoloration avant l'administration. Jeter le flacon si des particules et / ou une décoloration sont présentes.
- N'injectez pas la solution de Lutathera directement dans une autre solution intraveineuse.
- Confirmez la quantité de radioactivité de Lutathera livrée au patient avec un calibrateur de dose approprié avant et après chaque administration de Lutathera.
- Jetez tout produit médicinal ou déchet inutilisé conformément aux lois locales et fédérales.
Instructions d'administration
- Avant l'administration, rincez le cathéter intraveineux utilisé pour l'administration de Lutathera avec ≥ 10 ml de l'injection de chlorure de sodium 0,9% USP pour assurer la perméabilité et minimiser le risque d'extravasation. Gérer les cas d'extravasation conformément aux directives institutionnelles.
- La méthode de la méthode de gravité péristaltique de la méthode ou la méthode de la pompe de seringue peut être utilisée pour l'administration de la posologie recommandée. N'administrez pas Lutathera comme un bolus intraveineux.
- Lors de l'utilisation de la méthode de la gravité ou de la pompe péristaltique, infuser Lutathera directement à partir de son conteneur d'origine.
- Utilisez la pompe péristaltique ou la méthode de la pompe à seringue lors de l'administration d'une dose réduite de Lutathera après une modification posologique pour une réaction indésirable. Lorsque vous utilisez la méthode de gravité pour une dose réduite, ajustez la dose de Lutathera avant l'administration pour éviter la livraison d'un volume incorrect de Lutathera.
Méthodes d'administration intraveineuses
Instructions pour la méthode de gravité
- Insérez une aiguille de calibre 20 de 2,5 cm (aiguille courte) dans le flacon Lutathera et connectez-vous via un cathéter à 500 ml à 0,9% d'injection de chlorure de sodium USP (utilisé pour transporter la solution de Lutathera pendant la perfusion). Assurez-vous que l'aiguille courte ne touche pas la solution de Lutathera dans le flacon et ne connectez pas cette courte aiguille directement au patient. Ne permettez pas à l'injection de chlorure de sodium à 0,9% de s'écouler dans le flacon de Lutathera avant l'initiation de la perfusion de Lutathera et n'injectez pas la solution de Lutathera directement dans l'injection de chlorure de sodium à 0,9% USP.
- Insérez une deuxième aiguille de 9 cm de calibre 18 (aiguille longue) dans le flacon Lutathera en garantissant que cette longue aiguille se touche et est sécurisée au bas du flacon de Lutathera pendant toute la perfusion. Connectez l'aiguille longue au patient par un cathéter intraveineux qui est pré-rempli à 0,9% d'injection de chlorure de sodium USP et qui est utilisé pour la perfusion de Lutathera chez le patient.
- Utilisez une pince ou une pompe à perfusion pour réguler le débit de l'injection de chlorure de sodium à 0,9% USP via l'aiguille courte dans le flacon de Lutathera à un taux de 50 ml / heure à 100 ml / heure pendant 5 à 10 minutes, puis 200 ml / heure à 300 ml / heure pour 25 à 30 minutes (la solution à 0,9% du chlorure de sodium en entrant dans le vial au patient via le cathéter intraveineux connecté à l'aiguille longue sur une durée totale de 30 à 40 minutes).
- Pendant la perfusion, assurez-vous que le niveau de solution dans le flacon de Lutathera reste constant.
- Débranchez le flacon de la longue lignée d'aiguille et serrez la ligne USP d'injection de chlorure de sodium à 0,9% une fois que le niveau de radioactivité est stable pendant au moins cinq minutes.
- Suivez la perfusion avec une chasse intraveineuse de 25 ml de l'injection de chlorure de sodium à 0,9% USP à travers le cathéter intraveineux au patient.
Instructions pour la méthode de la pompe péristaltique
- Insérez une aiguille filtrée de 2,5 cm 20 de calibre (aiguille de ventilation courte) dans le flacon Lutathera. Assurez-vous que l'aiguille courte ne touche pas la solution de Lutathera dans le flacon et ne connectez pas cette courte aiguille directement au patient ou à la pompe péristaltique.
- Insérez une deuxième aiguille de 9 cm 18 cm (aiguille longue) dans le flacon de Lutathera en garantissant que la longue aiguille touche et est fixée au bas du flacon de Lutathera pendant toute la perfusion. Connectez l'aiguille longue et une injection de chlorure de sodium à 0,9% USP à une soupape d'arrêt à 3 voies via des tubes appropriés.
- Connectez la sortie de la vanne d'arrêt à 3 voies aux tubes installés du côté entrée de la pompe péristaltique selon les instructions du fabricant.
- Amenez la ligne en ouvrant la vanne d'arrêt à 3 voies et en pompant la solution de Lutathera à travers le tube jusqu'à ce qu'elle atteigne la sortie de la valve.
- Amenez le cathéter intraveineux qui sera connecté au patient en ouvrant la soupape de cock à 3 voies à l'injection de chlorure de sodium à 0,9% USP et en pompant l'injection de chlorure de sodium à 0,9% jusqu'à ce qu'elle quitte l'extrémité du tube du cathéter.
- Connectez le cathéter intraveineux amorcé au patient et définissez la valve à bille à 3 voies de telle sorte que la solution de Lutathera soit conforme à la pompe péristaltique.
- Infuser un volume approprié de solution de Lutathera sur une période de 30 à 40 minutes pour livrer la radioactivité souhaitée.
- Lorsque la radioactivité souhaitée de Lutathera a été livrée, arrêtez la pompe péristaltique, puis modifiez la position de la valve de cock d'arrêt à 3 voies afin que la pompe péristaltique soit conforme à l'injection de chlorure de sodium à 0,9% USP. Redémarrer la pompe péristaltique et infuser une rince par voie intraveineuse de 25 ml d'injection de chlorure de sodium à 0,9% par le cathéter intraveineux au patient.
Instructions pour la méthode de la pompe à seringue
- Retirez un volume approprié de solution de Lutathera pour livrer la radioactivité souhaitée en utilisant une seringue jetable équipée d'un bouclier de seringue et d'une aiguille stérile jetable qui est de 9 cm 18 cm (aiguille longue). Pour aider le retrait de la solution, une aiguille filtrée de 2,5 cm 20 (aiguille de ventilation courte) peut être utilisée pour réduire la résistance du flacon sous pression. Assurez-vous que l'aiguille courte ne touche pas la solution de Lutathera dans le flacon.
- Intégrez la seringue dans la pompe blindée et incluez une valve de coco stoploc à 3 voies entre la seringue et un cathéter intraveineux pré-rempli avec une injection de chlorure de sodium à 0,9% et utilisé pour l'administration de Lutathera au patient.
- Infuser un volume approprié de solution de Lutathera sur une période de 30 à 40 minutes pour livrer la radioactivité souhaitée.
- Lorsque la radioactivité de Lutathera souhaitée a été livrée, arrêtez la pompe à seringuer, puis modifiez la position de la valve de cock à 3 voies pour rincer la seringue avec 25 ml de l'injection de chlorure de sodium à 0,9% USP. Redémarrez la pompe à seringue.
- Une fois la chasse d'eau de la seringue terminée, effectuez une rinçage intraveineuse avec 25 ml d'injection de chlorure de sodium à 0,9% à travers le cathéter intraveineux au patient.
Radiation Dosimetry
La pénétration maximale du lutétium-177 dans les tissus est de 2,2 mm et la pénétration moyenne est de 0,67 mm. L'écart moyen et type (SD) des doses absorbées par les rayonnements estimées pour les adultes recevant de la Lutathera est présentée dans le tableau 3. La moyenne et le SD des doses absorbées par les rayonnements estimées pour les patients pédiatriques 12 ans et plus recevant Lutathera sont présentés dans le tableau 4.
Tableau 3. Dose absorbée par les rayonnements estimés pour Lutathera chez les adultes dans Netter-1
| (GY / GBIQ) (N = 20) | Dose absorbée calculée pour 4 × 7,4 GBQ (29.6 Activité cumulative GBQ) (Gy) | |||
| Organe | Signifier | SD | Signifier | SD |
| Surrénales | 0.037 | 0.016 | 1.1 | 0.5 |
| Cerveau | 0.027 | 0.016 | 0.8 | 0.5 |
| Seins | 0.027 | 0.015 | 0.8 | 0.4 |
| Mur de la vésicule biliaire | 0.042 | 0.019 | 1.2 | 0.6 |
| Mur de coeur | 0.032 | 0.015 | 0.9 | 0.4 |
| Rognons | 0.654 | 0.295 | 19.4 | 8.7 |
| Foiea | 0.299 | 0.226 | 8.9 | 6.7 |
| Bas-mur intestinal inférieur | 0.029 | 0.016 | 0.9 | 0.5 |
| Poumons | 0.031 | 0.015 | 0.9 | 0.4 |
| Muscle | 0.029 | 0.015 | 0.8 | 0.4 |
| Cellules ostéogéniques | 0.151 | 0.268 | 4.5 | 7.9 |
| Ovairesb | 0.031 | 0.013 | 0.9 | 0.4 |
| Pancréas | 0.038 | 0.016 | 1.1 | 0.5 |
| Moellec | 0.035 | 0.029 | 1.0 | 0.8 |
| Peau | 0.027 | 0.015 | 0.8 | 0.4 |
| Intestin grêle | 0.031 | 0.015 | 0.9 | 0.5 |
| Rate | 0.846 | 0.804 | 25.1 | 23.8 |
| Paroi de l'estomac | 0.032 | 0.015 | 0.9 | 0.5 |
| Testiculesd | 0.026 | 0.018 | 0.8 | 0.5 |
| Thymus | 0.028 | 0.015 | 0.8 | 0.5 |
| Thyroïde | 0.027 | 0.016 | 0.8 | 0.5 |
| Corps total | 0.052 | 0.027 | 1.6 | 0.8 |
| Supérieur grand intestin wal | 0.032 | 0.015 | 0.9 | 0.4 |
| Mur de vessie urinaire | 0.437 | 0.176 | 12.8 | 5.3 |
| Utérusb | 0.032 | 0.013 | 1.0 | 0.4 |
| aN = 18 (deux patients exclus parce que la dose absorbée par le foie a été biaisée par l'absorption des métastases hépatiques). bN = 9 (patientes uniquement). cMoelle dosimetry estimates were determined using blood radioactivity. dN = 11 (patients masculins uniquement). |
Tableau 4. Dose absorbée par le rayonnement estimé pour Lutathera chez les patients pédiatriques 12 ans et plus dans Netter-P
| (GY / GBIQ) (N = 8a) | Dose absorbée calculée pour 4 × 7,4 GBQ (29.6 Activité cumulative GBQ) (Gy) | |||
| Organe | Signifier | SD | Signifier | SD |
| Surrénales | 0.045 | 0.011 | 1.3 | 0.3 |
| Cerveau | 0.021 | 0.006 | 0.6 | 0.2 |
| Seinsb | 0.018 | 0.006 | 0.5 | 0.2 |
| Œsophage | 0.024 | 0.006 | 0.7 | 0.2 |
| Yeux | 0.021 | 0.006 | 0.6 | 0.2 |
| Mur de la vésicule biliaire | 0.031 | 0.011 | 0.9 | 0.3 |
| Mur de coeur | 0.024 | 0.006 | 0.7 | 0.2 |
| Rognons | 0.773 | 0.288 | 22.9 | 8.5 |
| Colon gauche | 0.265 | 0.081 | 7.8 | 2.4 |
| Foie | 0.216 | 0.231 | 6.4 | 6.8 |
| Poumons | 0.024 | 0.006 | 0.7 | 0.2 |
| Cellules ostéogéniques | 0.046 | 0.019 | 1.4 | 0.6 |
| Ovairesb | 0.026 | 0.007 | 0.8 | 0.2 |
| Pancréas | 0.027 | 0.007 | 0.8 | 0.2 |
| Pituitairec | 1.053 | 0.348 | 31.2 | 10.3 |
| Prostated | 0.026 | 0.006 | 0.8 | 0.2 |
| Droite | 0.272 | 0.085 | 8.0 | 2.5 |
| Moelle (blood)e | 0.027 | 0.005 | 0.8 | 0.2 |
| Moelle (image) e | 0.055 | 0.026 | 1.6 | 0.8 |
| Colon droit | 0.152 | 0.045 | 4.5 | 1.3 |
| Glandes salivaires | 0.036 | 0.017 | 1.1 | 0.5 |
| Intestin grêle | 0.046 | 0.013 | 1.3 | 0.4 |
| Rate | 0.733 | 0.304 | 21.7 | 9.0 |
| Paroi de l'estomac | 0.027 | 0.007 | 0.8 | 0.2 |
| Testiculesd | 0.021 | 0.005 | 0.6 | 0.2 |
| Thymus | 0.022 | 0.006 | 0.7 | 0.2 |
| Thyroïde | 0.022 | 0.006 | 0.6 | 0.2 |
| Corps total | 0.042 | 0.010 | 1.2 | 0.3 |
| Mur de vessie urinaire | 0.573 | 0.088 | 17.0 | 2.6 |
| Utérusb | 0.031 | 0.008 | 0.9 | 0.2 |
| aLes données sont regroupées pour 8 patients pédiatriques atteints de tumeurs positives pour les récepteurs de la somatostatine (SSTR), y compris 4 patients atteints de GEP-NETS. bN = 5 (patientes uniquement). cN = 7 (3 tumeurs GEP-NET 4 SSTR). Des estimations de la dosimétrie hypophysaire n'ont été effectuées que lorsque l'absorption hypophysaire a été clairement observée sur les images planes. En raison de la petite taille de la disponibilité de l'hypophyse pour la quantification uniquement à partir d'images planes et de l'interférence de l'activité dans les estimations de la muqueuse nasale peut être associée à une grande incertitude. L'estimation de la dose absorbée par la glande hypophysaire comprend les contributions à la dose absorbée de l'activité à l'hypophyse que seules les contributions à la dose d'autres tissus ne sont pas incluses. dN = 3 (patients masculins uniquement). eMoelle dosimetry estimates were determined either using blood radioactivity or by imaging et scaling of a representative region of the lumbar spine. |
Comment fourni
Formes et forces posologiques
Injection : 370 MBQ / ml (10 mc / ml) de lutetium lu 177 dotatate comme une solution claire et incolore à légèrement jaune dans un flacon à dose unique.
Stockage et manipulation
Lutathera Injection containing 370 MBq/mL (10 mCi/mL) of lutetium Lu 177 dotatate is a sterile preservative-free et clear colorless to slightly yellow solution for intravenous use supplied in a clear colorless Type I glass 30 mL single-dose vial containing 7.4 GBq (200 mCi) ± 10% of lutetium Lu 177 dotatate at the time of injection ( NDC
Le flacon de produit est enfermé dans un récipient à blindage de plomb ( NDC NDC
Conserver en dessous de 25 ° C (77 ° F). Ne congelez pas Lutathera. Conservez dans l'emballage d'origine pour protéger des rayonnements ionisants (blindage en plomb).
La durée de conservation est de 72 heures à compter de la date et de l'heure de l'étalonnage. Jeter de manière appropriée à 72 heures.
Le lutétium-177 pour Lutathera peut être préparé en utilisant deux sources différentes de nucléides stables (Lutetium-176 ou Ytterbium-176) entraînant une gestion différente des déchets. Consultez la documentation fournie avant d'utiliser Lutathera pour assurer une gestion appropriée des déchets.
Distribué par: Advanced Accelerator Applications USA Inc. Millburn NJ 07041. Révisé: novembre 2024
Effets secondaires pour Lutathera
Les effets indésirables cliniquement significatifs suivants sont décrits ailleurs dans l'étiquetage:
- Myélosuppression [voir Avertissements et précautions ]
- Syndrome myélodysplasique secondaire et leucémie [voir Avertissements et précautions ]
- Toxicité rénale [voir Avertissements et précautions ]
- Hépatotoxicité [voir Avertissements et précautions ]
- Réactions d'hypersensibilité [voir Avertissements et précautions ]
- Crise hormonale neuroendocrine [voir Avertissements et précautions ]
Expérience des essais cliniques
Étant donné que les essais cliniques sont menés dans des conditions de réaction indésirables très variables observées dans les essais cliniques d'un médicament ne peuvent pas être directement comparées aux taux dans les essais cliniques d'un autre médicament et ne peuvent pas refléter les taux observés dans la pratique.
Les données des avertissements et des précautions reflètent l'exposition à Lutathera chez 111 patients atteints de tumeurs neuroendocrines avancées avancées de l'intestin moyen (Netter-1). Des données de sécurité dans les avertissements et les précautions ont également été obtenues chez 22 patients supplémentaires dans une sous-étude pharmacocinétique non randomisée de Netter-1 et dans un sous-ensemble de patients (811 sur 1214) avec des tumeurs avancées pour les récepteurs somatostatines inscrits à Erasmus [ Avertissements et précautions ].
Population adulteNet-1
Les données de sécurité de Lutathera avec Octreotide ont été évaluées dans Netter-1 [voir Études cliniques ]. Patients with progressive somatostatin receptor-positive midgut carcinoid tumors received Lutathera 7.4 GBq (200 mCi) administered every 8 to 16 weeks concurrently with the recommended amino acid solution et with long-acting octreotide (30 mg administered by intramuscular injection within 24 hours of each Lutathera dose) (N = 111) or high-dose octreotide (defined as long-acting octreotide 60 mg by intramuscular injection every 4 weeks) (N = 112) [voir Études cliniques ]. Parmi les patients recevant de la Lutathera avec de l'octréotide, 79% ont reçu une dose cumulative> 22,2 GBQ (> 600 MCI) et 76% des patients ont reçu les quatre doses prévues. Six pour cent (6%) des patients ont nécessité une réduction de dose et 13% des patients ont abandonné Lutathera. Cinq patients ont abandonné la Lutathera pour les événements rénaux et 4 interrompus pour les toxicités hématologiques.
Le tableau 5 et le tableau 6 résument respectivement l'incidence des effets indésirables et des anomalies de laboratoire. Les effets indésirables les plus courants de grade 3-4 se produisant avec une plus grande fréquence chez les patients recevant de la Lutathera avec de l'octréotide par rapport aux patients ayant augmenté l'octréotide à haute dose (GGT) (20%) Vomitte (7%) et augmentation de l'aspartate d'aspartate Hyperglycémie et hypokaliémie (4% chacune) de l'aminotransférase (ALT) (4%).
Tableau 5. Réactions indésirables se produisant à une incidence plus élevée chez les patients recevant de la Lutathera avec un octreotide à longue durée d'action par rapport à l'octréotide à longue durée d'action à forte dose (entre la différence de bras ≥ 5% tous les gradations ou ≥ 2% de grades 3 à 4)a
| Réaction indésirablea | Lutathera with Long-acting Octreotide (30 mg) (N = 111) | Octreotide à action prolongée (60 mg) (N = 112) | ||
| Toutes les notes % | Grades 3-4 % | Toutes les notes % | Grades 3-4 % | |
| Troubles gastro-intestinaux | ||||
| Nausée | 65 | 5 | 12 | 2 |
| Vomissement | 53 | 7 | 10 | 0 |
| Douleurs abdominales | 26 | 3 | 19 | 3 |
| Diarrhée | 26 | 3 | 18 | 1 |
| Constipation | 10 | 0 | 5 | 0 |
| Troubles généraux | ||||
| Fatigue | 38 | 1 | 26 | 2 |
| Œdème périphérique | 16 | 0 | 9 | 1 |
| Pyrexie | 8 | 0 | 3 | 0 |
| Métabolisme et troubles nutritionnels | ||||
| Diminution de l'appétit | 21 | 0 | 11 | 3 |
| Troubles du système nerveux | ||||
| Mal de tête | 17 | 0 | 5 | 0 |
| Vertiges | 17 | 0 | 8 | 0 |
| Dysgeusie | 8 | 0 | 2 | 0 |
| Troubles vasculaires | ||||
| Bouffée | 14 | 1 | 9 | 0 |
| Hypertension | 12 | 2 | 7 | 2 |
| Troubles du tissu musculo-squelettique et conjonctif | ||||
| Maux de dos | 13 | 2 | 10 | 0 |
| Douleur à l'extrémité | 11 | 0 | 5 | 0 |
| Myalgie | 5 | 0 | 0 | 0 |
| Douleurs au cou | 5 | 0 | 0 | 0 |
| Troubles rénaux et urinaires | ||||
| Insuffisance rénaleb | 13 | 3 | 4 | 1 |
| Radiation-related urinary tract adverse reactionsc | 8 | 0 | 3 | 0 |
| Troubles psychiatriques | ||||
| Anxiété | `12 | 1 | 5 | 0 |
| Peau et subcutaneous tissue disorders | ||||
| Alopécie | 12 | 0 | 2 | 0 |
| Troubles thoraciques et médiastinaux respiratoires | ||||
| Toux | 11 | 1 | 6 | 0 |
| Troubles cardiaques | ||||
| Fibrillation auriculaire | 5 | 1 | 0 | 0 |
| aNational Cancer Institute Critères de terminologie commune pour les événements indésirables (CTCAE) Version 4.03. Affiche uniquement les effets indésirables survenant à une incidence plus élevée chez les patients traités par Lutathera [entre la différence de bras ≥ 5% (tous les grades) ou ≥ 2% (grades 3-4)]. bComprend les termes: le taux de filtration glomérulaire a diminué les lésions rénales aiguës de l'insuffisance prérénale aiguë azotémie trouble rénal insuffisance rénale altération rénale. cComprend les termes: dysurie miction d'urgence nocturia pollakiuria colique rénale douleur rénale des voies urinaires douleur et incontinence urinaire. |
Tableau 6. Anomalies de laboratoire se produisant à une incidence plus élevée chez les patients recevant de la Lutathera avec de l'octréotide à action longue par rapport à l'octréotide à longue durée d'action longue (entre la différence de bras ≥ 5% tous les gradations ou ≥ 2% de grades 3-4)AB
| Anomalie du laboratoireb | Lutathera with Long-acting Octreotide (30 mg) (N = 111) | Octreotide à action prolongée (60 mg) (N = 112) | ||
| Toutes les notes % | Grades 3-4 % | Toutes les notes % | Grades 3-4 % | |
| Hématologie | ||||
| Lymphopénie | 90 | 44 | 39 | 5 |
| Anémie | 81 | 0 | 55 | 1 |
| Leucopénie | 55 | 2 | 20 | 0 |
| Thrombocytopénie | 53 | 1 | 17 | 0 |
| Neutropénie | 26 | 3 | 11 | 0 |
| Rénal / métabolique | ||||
| La créatinine a augmenté | 85 | 1 | 73 | 0 |
| Hyperglycémie | 82 | 4 | 67 | 2 |
| Hyperuricémie | 34 | 6 | 30 | 6 |
| Hypocalcémie | 32 | 0 | 14 | 0 |
| Hypokaliémie | 26 | 4 | 21 | 2 |
| Hyperkaliémie | 19 | 0 | 11 | 0 |
| Hypernatrémie | 17 | 0 | 7 | 0 |
| Hypoglycémie | 15 | 0 | 8 | 0 |
| Hépatique | ||||
| GGT a augmenté | 66 | 20 | 67 | 16 |
| La phosphatase alcaline a augmenté | 65 | 5 | 55 | 9 |
| AST a augmenté | 50 | 5 | 35 | 0 |
| Alt a augmenté | 43 | 4 | 34 | 0 |
| La bilirubine sanguine a augmenté | 30 | 2 | 28 | 0 |
| aLes valeurs sont la pire note observée après randomisation. bNational Cancer Institute Critères de terminologie commune pour les événements indésirables (CTCAE) Version 4.03. Affiche uniquement les anomalies de laboratoire survenant à une incidence plus élevée chez les patients traités par Lutathera [entre la différence de bras ≥ 5% (tous les grades) ou ≥ 2% (grades 3-4)]. |
Érasmus
Les données de sécurité sont disponibles auprès de 1214 patients d'Erasmus et d'essai international en ouvert un seul bras unique de patients atteints de tumeurs positives par les récepteurs de la somatostatine (neuroendocrine et autres primaires). Les patients ont reçu Lutathera 7,4 GBQ (200 MCI) administrés toutes les 6 à 13 semaines avec ou sans octreotide. Un examen rétrospectif des dossiers médicaux a été effectué sur un sous-ensemble de 811 patients pour documenter les effets indésirables graves. Quatre-vingt-un (81%) pour cent des patients du sous-ensemble ont reçu une dose cumulée ≥ 22,2 GBQ (≥ 600 MCI). Avec un temps de suivi médian de plus de 4 ans, les taux suivants de réactions indésirables graves ont été signalés: Syndrome myélodysplasique (2%) La leucémie aiguë (1%) Insuffisance rénale (2%) Hypotension (1%) Échec cardiaque (2%) infarctus du myocarde (1%) et crurisation hormonale neuroendocrine (1%).
Population pédiatriqueNette-p
Les données de sécurité sont disponibles auprès de 9 patients pédiatriques de Netter-P (NCT04711135) un essai international en ouvert à bras unique multicentrique de patients atteints de tumeurs positives pour les récepteurs somatostatine, dont 4 patients atteints de NIF GEP. Les patients ont reçu Lutathera 7,4 GBQ (200 MCI) administrés toutes les 8 semaines simultanément avec la solution d'acides aminés recommandée. Les effets indésirables observés dans Netter-P étaient similaires à ceux observés chez les adultes traités avec Lutathera.
Expérience de commercialisation de la poste
Les effets indésirables suivants ont été identifiés lors de l'utilisation post-approbation de Lutathera. Étant donné que ces réactions sont rapportées volontairement d'une population de taille incertaine, il n'est pas toujours possible d'estimer de manière fiable leur fréquence ou d'établir une relation causale à l'exposition au médicament.
- Troubles du système immunitaire: Réactions d'hypersensibilité, y compris l'œdème de l'angio
Interactions médicamenteuses pour Lutathera
Analogues de somatostatine
La somatostatine et ses analogues se lient de manière compétitive aux récepteurs de la somatostatine et peuvent interférer avec l'efficacité de Lutathera. Interrompre les analogues de la somatostatine à action prolongée au moins 4 semaines et l'octréotide à action courte au moins 24 heures avant chaque dose de Lutathera. Administrer l'octréotide à action courte et longue pendant le traitement de Lutathera comme recommandé [voir Posologie et administration ].
Glucocorticoïdes
Glucocorticoïdes can induce down-regulation of subtype 2 somatostatin receptors (SSTR2). Avoid repeated administration of high doses of glucocorticoids during treatment with Lutathera.
Avertissements pour Lutathera
Inclus dans le cadre du 'PRÉCAUTIONS' Section
Précautions pour Lutathera
Risque de l'exposition aux radiations
Lutathera contributes to a patient’s overall long-term cumulative radiation exposure. Long-term cumulative radiation exposure is associated with an increased risk for cancer. These risks of radiation associated with the use of Lutathera are greater in pediatric patients than in adults [voir Utiliser dans des populations spécifiques ].
Radiation can be detected in the urine for up to 30 days following Lutathera administration. Minimize radiation exposure to patients medical personnel et household contacts during et after treatment with Lutathera consistent with institutional good radiation safety practices patient management procedures Nuclear Regulatory Commission patient-release guidance et instructions to the patient for follow-up radiation protection at home [voir Posologie et administration Pharmacologie clinique ].
Myélosuppression
Dans la myélosuppression Netter-1 s'est produit plus fréquemment chez les patients recevant de la Lutathera avec un octreotide à action prolongée par rapport aux patients recevant de l'octréotide à longue durée d'action longue (tous les grades / grade 3 ou 4): anémie (81% / 0) contre (54% / 1%); thrombocytopénie (53% / 1%) contre (17% / 0); et neutropénie (26% / 3%) par rapport (11% / 0). En plaquette Netter-1, le nadir s'est produit à une médiane de 5,1 mois après la première dose. Sur les 59 patients qui ont développé une thrombocytopénie, 68% ont eu une récupération des plaquettes à des niveaux de base ou normaux. Le délai médian de récupération des plaquettes était de 2 mois. Fifteen of the nineteen patients in whom platelet recovery was not documented had post-nadir platelet counts. Parmi ces 15 patients 5 se sont améliorés de grade 1 9 à grade 2 et 1 à grade 3.
Surveiller le nombre de cellules sanguines. Retenir la dose de réduction de la dose ou interrompre de façon permanente Lutathera en fonction de la gravité de la myélosuppression [voir Posologie et administration ].
Syndrome myélodysplasique secondaire et leucémie
Dans Netter-1 avec un temps de suivi médian de 76 mois dans l'étude principale, le syndrome myélodysplasique (MDS) a été signalé chez 2,3% des patients recevant de la Lutathera avec de l'octréotide à longue durée d'action par rapport à aucun patient ne recevant une octréotide à longue durée d'action longue.
Chez Erasmus, 16 patients (2,0%) ont développé des MD et 4 (0,5%) ont développé une leucémie aiguë. Le délai médian de début était de 29 mois (9 à 45 mois) pour les MD et 55 mois (32 à 125 mois) pour la leucémie aiguë.
Toxicité rénale
Chez Erasmus 8 patients (<1%) developed renal failure 3 to 36 months following Lutathera. Two of these patients had underlying renal impairment or risk factors for renal failure (e.g. diABetes or hypertension) et required dialysis.
Administrer la solution d'acide aminé recommandée avant et après Lutathera [voir Posologie et administration ] pour diminuer la réabsorption du lutetium lu 177 dotaté à travers les tubules proximaux et diminuer la dose de rayonnement aux reins. Conseiller aux patients d'hydrater et d'uriner fréquemment avant le jour et le lendemain de l'administration de Lutathera.
Surveillez la créatinine sérique et la clairance de la créatinine calculée. Retenir la dose de réduction de la dose ou interrompre de façon permanente Lutathera en fonction de la gravité de la toxicité rénale [voir Posologie et administration ].
Les patients présentant une déficience rénale de base peuvent être à un risque accru de toxicité en raison de l'augmentation de l'exposition aux radiations [voir Utiliser dans des populations spécifiques ].
Hépatotoxicité
Chez Erasmus 2 patients (<1%) were reported to have hepatic tumor hemorrhage edema or necrosis with one patient experiencing intrahepatic congestion et cholestasis. Patients with hepatic metastasis may be at increased risk of hepatotoxicity due to radiation exposure.
Surveiller les transaminases à bilirubine sérique albumine et le rapport international normalisé (INR) pendant le traitement. Retenir la dose de réduction de la dose ou interrompre de façon permanente Lutathera en fonction de la gravité de l'hépatotoxicité [voir Posologie et administration ].
Réactions d'hypersensibilité
Des réactions d'hypersensibilité, y compris l'œdème de l'angio Effets indésirables ]. Monitor patients closely for signs et symptoms of hypersensitivity reactions including anaphylaxis during et following Lutathera administration for a minimum of 2 hours in a setting where cardiopulmonary resuscitation medication et equipment are availABle. Discontinue the infusion upon the first observation of any signs or symptoms consistent with a severe hypersensitivity reaction et initiate appropriate therapy.
Prémédicate des patients ayant des antécédents de réactions d'hypersensibilité de grade 1 ou 2 à Lutathera avant les doses ultérieures [voir Posologie et administration ]. Permanently discontinue Lutathera in patients who experience Grade 3 or 4 hypersensitivity reactions [voir Posologie et administration ].
Crise hormonale neuroendocrine
Des crises hormonales neuroendocrines se manifestant avec du bronchospasme de diarrhée et une hypotension se sont produites dans <1% of patients in Érasmus et typically occurred during or within 24 hours following the initial Lutathera dose. Two (< 1%) patients were reported to have hypercalcemia.
Surveiller les patients pour le rinçage de la diarrhée hypotension bronchoconstriction ou d'autres signes et symptômes de libération hormonale liée à la tumeur. Administrer des analogues de somatostatine intraveineux fluides corticostéroïdes et électrolytes comme indiqué.
Toxicité embryo-fœtale
Sur la base de son mécanisme d'action, Lutathera peut causer des dommages fœtaux lorsqu'il est administré à une femme enceinte [voir Pharmacologie clinique ]. There are no availABle data on Lutathera use in pregnant women. No animal studies using lutetium Lu 177 dotatate have been conducted to evaluate its effect on female reproduction et embryo-fetal development; however radioactive emissions including those from Lutathera can cause fetal harm.
Vérifiez le statut de grossesse des femmes de potentiel reproducteur avant de lancer Lutathera [voir Posologie et administration ].
Conseiller les femmes enceintes du risque potentiel pour un fœtus. Conseiller les femmes de potentiel de reproduction à utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 7 mois après la dernière dose. Conseiller les hommes avec des partenaires féminins de potentiel reproducteur pour utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 4 mois après la dernière dose [voir Utiliser dans des populations spécifiques ].
Risque d'infertilité
Lutathera may cause infertility in males et females. La dose cumulative recommandée de 29,6 GBQ de Lutathera entraîne une dose absorbée par le rayonnement aux testicules et aux ovaires dans la plage où une infertilité temporaire ou permanente peut être attendue après la radiothérapie du faisceau externe [voir Posologie et administration Utiliser dans des populations spécifiques ].
Toxicologie non cliniqueCarcinogenèse Mutagenèse A trouble de la fertilité
Les études de cancérogénicité et de mutagénicité n'ont pas été menées avec le lutetium lu 177 dotatate; Cependant, le rayonnement est un cancérogène et un mutagène.
Aucune étude animale n'a été menée pour déterminer les effets du lutetium lu 177 dotaté sur la fertilité.
Utilisation dans une population spécifiqueGrossesseRésumé des risques
Résumé des risques
Sur la base de son mécanisme d'action, Lutathera peut causer des dommages fœtaux lorsqu'il est administré à une femme enceinte [voir Pharmacologie clinique ]. There are no availABle data on Lutathera use in pregnant women. No animal studies using lutetium Lu 177 dotatate have been conducted to evaluate its effect on female reproduction et embryo-fetal development; however radioactive emissions including those from Lutathera can cause fetal harm. Advise pregnant women of the potential risk to a fetus.
Dans la population générale américaine, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est respectivement de 2% à 4% et 15% à 20%.
LactationRésumé des risques
Il n'y a pas de données sur la présence du lutetium Lu 177 dotaTate dans le lait maternel ou ses effets sur la production d'enfant allaitée ou de lait. Aucune étude de lactation chez les animaux n'a été menée. En raison du risque potentiel de réactions indésirables graves chez les enfants allaités conseille aux femmes de ne pas allaiter pendant le traitement par Lutathera et pendant 2,5 mois après la dernière dose.
Femmes et mâles de potentiel reproducteur
Sur la base du mécanisme d'action, Lutathera peut causer des dommages fœtaux lorsqu'il est administré à une femme enceinte [voir Grossesse ].
Grossesse Testing
Vérifiez le statut de grossesse des femmes de potentiel reproducteur avant de lancer Lutathera [voir Grossesse ].
Contraception
Femelles
Conseiller les femmes de potentiel de reproduction à utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 7 mois après la dernière dose.
Hommes
Conseiller les hommes avec des partenaires féminins de potentiel reproducteur pour utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 4 mois après la dernière dose [voir Pharmacologie clinique Toxicologie non clinique ].
Infertilité
La dose cumulative recommandée de 29,6 GBQ de Lutathera entraîne une dose absorbée par le rayonnement aux testicules et aux ovaires dans la plage où une infertilité temporaire ou permanente peut être attendue après la radiothérapie du faisceau externe [voir Posologie et administration ].
Usage pédiatriqueTumeurs neuroendocrines gastroentéroprantes pour les récepteurs de la somatostatine positifs
La sécurité et l'efficacité de la Lutathera ont été établies chez les patients pédiatriques de 12 ans et plus avec une neuroendocrine gastroentéropancréatique (GEP-NET) gastro-uentéropancréatique (GEP-NET). L'utilisation de Lutathera pour cette indication est étayée par des preuves d'une étude adéquate et bien contrôlée de Lutathera chez des adultes atteints de données pharmacocinétiques et de dosimétrie de sécurité supplémentaires chez les patients pédiatriques âgés de 12 ans et plus avec des tumoraux de somatostatine-récepteur-positives, dont 4 patients pédiatriques atteints de NET GEP [voir les récepteurs somatostatines, dont 4 patients pédiatriques atteints de NET GEP [voir les récepteurs somatostatines, dont 4 patients pédiatriques atteints de GEP-NETS [voir Somatostatine Receptor-Positive Tumers, dont 4 patients pédiatriques atteints de NET GEP [voir Somatostatine Receptor Effets indésirables Pharmacologie clinique et Études cliniques ].
Les risques d'exposition aux radiations associés à Lutathera sont plus élevés chez les patients pédiatriques que chez les patients adultes en raison d'une espérance de vie plus longue. Un suivi continu est recommandé pour l'évaluation des effets à long terme.
Il n'y avait pas de différence cliniquement pertinente dans l'exposition au lutetium LU 177 DotaTate chez les patients pédiatriques âgés de 13 à 16 ans par rapport aux patients adultes [voir Pharmacologie clinique ].
Le profil pharmacocinétique et la sécurité de la Lutathera chez les patients pédiatriques de 12 ans et plus avec une insuffisance rénale de base n'ont pas été étudiés.
La sécurité et l'efficacité de la Lutathera n'ont pas été établies chez des patients pédiatriques de moins de 12 ans atteints de GEP-NET GEP-NET pour les récepteurs de la somatostatine.
Utilisation gériatrique
Sur les 1325 patients traités par Lutathera dans des essais cliniques, 438 patients (33%) étaient de 65 ans et plus. Aucune différence globale de sécurité ou d'efficacité n'a été observée entre les patients plus âgés et plus jeunes.
Trouble rénal
Aucun ajustement de dose n'est recommandé pour les patients atteints de base de base à une insuffisance rénale de base (clairance de créatinine de 30 à 89 ml / min par formule de cockcroft-gault). Cependant, les patients présentant une altération rénale légère ou modérée de base peuvent être plus à risque de toxicité, y compris la toxicité rénale en raison de l'augmentation de l'exposition aux radiations. Effectuer des évaluations plus fréquentes de la fonction rénale chez les patients atteints de déficience de base légère à modérée. Le profil pharmacocinétique et la sécurité de Lutathera chez les patients atteints de déficience rénale grave de base (autorisation de créatinine <30 mL/min by Cockcroft-Gault formula) or end-stage renal disease have not been studied [voir Avertissements et précautions ].
Avertissements pour Lutathera
Inclus dans le cadre du 'PRÉCAUTIONS' Section
Précautions pour Lutathera
Risque de l'exposition aux radiations
Lutathera contributes to a patient’s overall long-term cumulative radiation exposure. Long-term cumulative radiation exposure is associated with an increased risk for cancer. These risks of radiation associated with the use of Lutathera are greater in pediatric patients than in adults [voir Utiliser dans des populations spécifiques ].
Radiation can be detected in the urine for up to 30 days following Lutathera administration. Minimize radiation exposure to patients medical personnel et household contacts during et after treatment with Lutathera consistent with institutional good radiation safety practices patient management procedures Nuclear Regulatory Commission patient-release guidance et instructions to the patient for follow-up radiation protection at home [voir Posologie et administration Pharmacologie clinique ].
Myélosuppression
Dans la myélosuppression Netter-1 s'est produit plus fréquemment chez les patients recevant de la Lutathera avec un octreotide à action prolongée par rapport aux patients recevant de l'octréotide à longue durée d'action longue (tous les grades / grade 3 ou 4): anémie (81% / 0) contre (54% / 1%); thrombocytopénie (53% / 1%) contre (17% / 0); et neutropénie (26% / 3%) par rapport (11% / 0). En plaquette Netter-1, le nadir s'est produit à une médiane de 5,1 mois après la première dose. Sur les 59 patients qui ont développé une thrombocytopénie, 68% ont eu une récupération des plaquettes à des niveaux de base ou normaux. Le délai médian de récupération des plaquettes était de 2 mois. Fifteen of the nineteen patients in whom platelet recovery was not documented had post-nadir platelet counts. Parmi ces 15 patients 5 se sont améliorés de grade 1 9 à grade 2 et 1 à grade 3.
Surveiller le nombre de cellules sanguines. Retenir la dose de réduction de la dose ou interrompre de façon permanente Lutathera en fonction de la gravité de la myélosuppression [voir Posologie et administration ].
Syndrome myélodysplasique secondaire et leucémie
Dans Netter-1 avec un temps de suivi médian de 76 mois dans l'étude principale, le syndrome myélodysplasique (MDS) a été signalé chez 2,3% des patients recevant de la Lutathera avec de l'octréotide à longue durée d'action par rapport à aucun patient ne recevant une octréotide à longue durée d'action longue.
Chez Erasmus, 16 patients (2,0%) ont développé des MD et 4 (0,5%) ont développé une leucémie aiguë. Le délai médian de début était de 29 mois (9 à 45 mois) pour les MD et 55 mois (32 à 125 mois) pour la leucémie aiguë.
Toxicité rénale
Chez Erasmus 8 patients (<1%) developed renal failure 3 to 36 months following Lutathera. Two of these patients had underlying renal impairment or risk factors for renal failure (e.g. diABetes or hypertension) et required dialysis.
Administrer la solution d'acide aminé recommandée avant et après Lutathera [voir Posologie et administration ] pour diminuer la réabsorption du lutetium lu 177 dotaté à travers les tubules proximaux et diminuer la dose de rayonnement aux reins. Conseiller aux patients d'hydrater et d'uriner fréquemment avant le jour et le lendemain de l'administration de Lutathera.
Surveillez la créatinine sérique et la clairance de la créatinine calculée. Retenir la dose de réduction de la dose ou interrompre de façon permanente Lutathera en fonction de la gravité de la toxicité rénale [voir Posologie et administration ].
Les patients présentant une déficience rénale de base peuvent être à un risque accru de toxicité en raison de l'augmentation de l'exposition aux radiations [voir Utiliser dans des populations spécifiques ].
Hépatotoxicité
Chez Erasmus 2 patients (<1%) were reported to have hepatic tumor hemorrhage edema or necrosis with one patient experiencing intrahepatic congestion et cholestasis. Patients with hepatic metastasis may be at increased risk of hepatotoxicity due to radiation exposure.
Surveiller les transaminases à bilirubine sérique albumine et le rapport international normalisé (INR) pendant le traitement. Retenir la dose de réduction de la dose ou interrompre de façon permanente Lutathera en fonction de la gravité de l'hépatotoxicité [voir Posologie et administration ].
Réactions d'hypersensibilité
Des réactions d'hypersensibilité, y compris l'œdème de l'angio Effets indésirables ]. Monitor patients closely for signs et symptoms of hypersensitivity reactions including anaphylaxis during et following Lutathera administration for a minimum of 2 hours in a setting where cardiopulmonary resuscitation medication et equipment are availABle. Discontinue the infusion upon the first observation of any signs or symptoms consistent with a severe hypersensitivity reaction et initiate appropriate therapy.
Prémédicate des patients ayant des antécédents de réactions d'hypersensibilité de grade 1 ou 2 à Lutathera avant les doses ultérieures [voir Posologie et administration ]. Permanently discontinue Lutathera in patients who experience Grade 3 or 4 hypersensitivity reactions [voir Posologie et administration ].
Crise hormonale neuroendocrine
Des crises hormonales neuroendocrines se manifestant avec du bronchospasme de diarrhée et une hypotension se sont produites dans <1% of patients in Érasmus et typically occurred during or within 24 hours following the initial Lutathera dose. Two (< 1%) patients were reported to have hypercalcemia.
Surveiller les patients pour le rinçage de la diarrhée hypotension bronchoconstriction ou d'autres signes et symptômes de libération hormonale liée à la tumeur. Administrer des analogues de somatostatine intraveineux fluides corticostéroïdes et électrolytes comme indiqué.
Toxicité embryo-fœtale
Sur la base de son mécanisme d'action, Lutathera peut causer des dommages fœtaux lorsqu'il est administré à une femme enceinte [voir Pharmacologie clinique ]. There are no availABle data on Lutathera use in pregnant women. No animal studies using lutetium Lu 177 dotatate have been conducted to evaluate its effect on female reproduction et embryo-fetal development; however radioactive emissions including those from Lutathera can cause fetal harm.
Vérifiez le statut de grossesse des femmes de potentiel reproducteur avant de lancer Lutathera [voir Posologie et administration ].
Conseiller les femmes enceintes du risque potentiel pour un fœtus. Conseiller les femmes de potentiel de reproduction à utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 7 mois après la dernière dose. Conseiller les hommes avec des partenaires féminins de potentiel reproducteur pour utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 4 mois après la dernière dose [voir Utiliser dans des populations spécifiques ].
Risque d'infertilité
Lutathera may cause infertility in males et females. La dose cumulative recommandée de 29,6 GBQ de Lutathera entraîne une dose absorbée par le rayonnement aux testicules et aux ovaires dans la plage où une infertilité temporaire ou permanente peut être attendue après la radiothérapie du faisceau externe [voir Posologie et administration Utiliser dans des populations spécifiques ].
Toxicologie non cliniqueCarcinogenèse Mutagenèse A trouble de la fertilité
Les études de cancérogénicité et de mutagénicité n'ont pas été menées avec le lutetium lu 177 dotatate; Cependant, le rayonnement est un cancérogène et un mutagène.
Aucune étude animale n'a été menée pour déterminer les effets du lutetium lu 177 dotaté sur la fertilité.
Utilisation dans une population spécifiqueGrossesseRésumé des risques
Résumé des risques
Sur la base de son mécanisme d'action, Lutathera peut causer des dommages fœtaux lorsqu'il est administré à une femme enceinte [voir Pharmacologie clinique ]. There are no availABle data on Lutathera use in pregnant women. No animal studies using lutetium Lu 177 dotatate have been conducted to evaluate its effect on female reproduction et embryo-fetal development; however radioactive emissions including those from Lutathera can cause fetal harm. Advise pregnant women of the potential risk to a fetus.
Dans la population générale américaine, le risque de fond estimé de malformations congénitales majeures et de fausse couche dans les grossesses cliniquement reconnues est respectivement de 2% à 4% et 15% à 20%.
LactationRésumé des risques
Il n'y a pas de données sur la présence du lutetium Lu 177 dotaTate dans le lait maternel ou ses effets sur la production d'enfant allaitée ou de lait. Aucune étude de lactation chez les animaux n'a été menée. En raison du risque potentiel de réactions indésirables graves chez les enfants allaités conseille aux femmes de ne pas allaiter pendant le traitement par Lutathera et pendant 2,5 mois après la dernière dose.
Femmes et mâles de potentiel reproducteur
Sur la base du mécanisme d'action, Lutathera peut causer des dommages fœtaux lorsqu'il est administré à une femme enceinte [voir Grossesse ].
Grossesse Testing
Vérifiez le statut de grossesse des femmes de potentiel reproducteur avant de lancer Lutathera [voir Grossesse ].
Contraception
Femelles
Conseiller les femmes de potentiel de reproduction à utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 7 mois après la dernière dose.
Hommes
Conseiller les hommes avec des partenaires féminins de potentiel reproducteur pour utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 4 mois après la dernière dose [voir Pharmacologie clinique Toxicologie non clinique ].
Infertilité
La dose cumulative recommandée de 29,6 GBQ de Lutathera entraîne une dose absorbée par le rayonnement aux testicules et aux ovaires dans la plage où une infertilité temporaire ou permanente peut être attendue après la radiothérapie du faisceau externe [voir Posologie et administration ].
Usage pédiatriqueTumeurs neuroendocrines gastroentéroprantes pour les récepteurs de la somatostatine positifs
La sécurité et l'efficacité de la Lutathera ont été établies chez les patients pédiatriques 12 ans et plus avec des tumeurs neuroendocrines gastroentéropancréatiques (GEP-NETS gastroentéropancréatiques (GEP-NET). L'utilisation de la Flutathera pour cette indication est étayée par des preuves d'une étude adéquate et bien contrôlée de la Flutathera chez des adultes atteints de données pharmacocinétiques et de dosimétrie de sécurité supplémentaires chez les patients pédiatriques âgés de 12 ans et plus avec des tumeurs de somatostatine-récepteur-positives, dont 4 patients pédiatriques atteints de NET GEP [voir les récepteurs somatostas Effets indésirables Pharmacologie clinique Études cliniques ].
Les risques d'exposition aux radiations associés à Lutathera sont plus élevés chez les patients pédiatriques que chez les patients adultes en raison d'une espérance de vie plus longue. Un suivi continu est recommandé pour l'évaluation des effets à long terme.
Il n'y avait pas de différence cliniquement pertinente dans l'exposition au lutetium LU 177 DotaTate chez les patients pédiatriques âgés de 13 à 16 ans par rapport aux patients adultes [voir Pharmacologie clinique ].
Le profil pharmacocinétique et la sécurité de la Lutathera chez les patients pédiatriques de 12 ans et plus avec une insuffisance rénale de base n'ont pas été étudiés.
La sécurité et l'efficacité de la Lutathera n'ont pas été établies chez des patients pédiatriques de moins de 12 ans atteints de GEP-NET GEP-NET pour les récepteurs de la somatostatine.
Utilisation gériatrique
Sur les 1325 patients traités par Lutathera dans des essais cliniques, 438 patients (33%) étaient de 65 ans et plus. Aucune différence globale de sécurité ou d'efficacité n'a été observée entre les patients plus âgés et plus jeunes.
Trouble rénal
Aucun ajustement de dose n'est recommandé pour les patients atteints de base de base à une insuffisance rénale de base (clairance de créatinine de 30 à 89 ml / min par formule de cockcroft-gault). Cependant, les patients présentant une altération rénale légère ou modérée de base peuvent être plus à risque de toxicité, y compris la toxicité rénale en raison de l'augmentation de l'exposition aux radiations. Effectuer des évaluations plus fréquentes de la fonction rénale chez les patients atteints de déficience de base légère à modérée. Le profil pharmacocinétique et la sécurité de Lutathera chez les patients atteints de déficience rénale grave de base (autorisation de créatinine <30 mL/min by Cockcroft-Gault formula) or end-stage renal disease have not been studied [voir Avertissements et précautions ].
Hépatique Impairment
Aucun ajustement de dose n'est recommandé pour les patients atteints de déficience hépatique légère ou modérée de base. Le profil pharmacocinétique et la sécurité de Lutathera chez les patients présentant une déficience hépatique sévère de base (bilirubine totale> 3 fois la limite supérieure de la normale quel que soit le niveau AST) n'ont pas été étudiées.
Hépatique Impairment
Aucun ajustement de dose n'est recommandé pour les patients atteints de déficience hépatique légère ou modérée de base. Le profil pharmacocinétique et la sécurité de Lutathera chez les patients présentant une déficience hépatique sévère de base (bilirubine totale> 3 fois la limite supérieure de la normale quel que soit le niveau AST) n'ont pas été étudiées.
Informations sur la surdose pour Lutathera
Aucune information fournie
Contre-indications pour Lutathera
Aucun.
Pharmacologie clinique for Lutathera
Mécanisme d'action
Le lutetium Lu 177 dotatate se lie aux récepteurs de la somatostatine avec une affinité la plus élevée pour les récepteurs de la somatostatine du sous-type 2 (SSTR2). En se liant aux cellules exprimant les récepteurs de la somatostatine, y compris les tumeurs malignes des récepteurs de la somatostatine positive, le composé est internalisé. L'émission bêta-minus du lutétium-177 induit des dommages cellulaires par la formation de radicaux libres dans les cellules positives par les récepteurs de la somatostatine et dans les cellules voisines.
Pharmacodynamique
Le lutetium Lu 177 Les relations avec la réponse à l'exposition dotaTate et l'évolution temporelle de la réponse pharmacodynamique sont inconnues.
Électrophysiologie cardiaque
La capacité de Lutathera à prolonger l'intervalle QTC à la dose recommandée a été évaluée dans une étude ouverte chez 20 patients atteints de tumeurs carcinoïdes moyennes de l'intestin moyen positives par les récepteurs de la somatostatine. Aucun changement important dans l'intervalle QTC moyen (c'est-à-dire> 20 ms) n'a été détecté.
Pharmacocinétique
La pharmacocinétique (PK) du lutetium Lu 177 dotatate a été caractérisée chez les patients atteints de tumeurs neuroendocrines progressives des récepteurs de la somatostatine positive.
L'exposition sanguine moyenne (superficie sous la courbe) du lutetium Lu 177 dotate à la dose recommandée est de 41 ng.h / ml [coefficient de variation (CV) 36%]. La concentration sanguine maximale moyenne (CMAX) pour le lutetium Lu 177 dotatate est de 10 ng / ml (CV 50%) qui s'est généralement produite à la fin de la perfusion de Lutathera.
Distribution
Le volume moyen de distribution (VZ) pour le lutetium Lu 177 dotaTate est de 460 L (CV 54%).
Le lutetium Lutetium LU 175 non radioactif est à 43% lié aux protéines plasmatiques humaines.
Dans les 4 heures suivant l'administration, le lutetium Lu 177 Dotatate se répartit dans les lésions tumorales de la rate du foie et dans certains patients, pituitaire et thyroïde. La co-administration des acides aminés a réduit la dose de rayonnement médiane aux reins de 47% (34% à 59%) et a augmenté la clairance sanguine en phase bêta moyenne du lutetium lu 177 dotaTate de 36%.
Élimination
La clairance moyenne (CL) est de 4,5 l / h (CV 31%) et la demi-vie terminale moyenne est de 71 (± 28) heures pour le lutétium 177 dotaté.
Métabolisme
effets secondaires de la prednisone et du cœur
Le lutetium lu 177 dotatate ne subit pas de métabolisme hépatique.
Excrétion
Le lutetium Lu 177 dotatate est principalement éliminé de manière rénale avec une excrétion cumulée de 44% dans les 5 heures 58% dans les 24 heures et 65% dans les 48 heures suivant l'administration de Lutathera. L'élimination prolongée du lutetium Lu 177 dotaTate dans l'urine est attendue; Cependant, sur la base de la demi-vie du lutetium-177 et de la demi-vie terminale du lutetium lu 177 dotatate supérieur à 99% de la radioactivité administrée sera éliminée dans les 14 jours suivant l'administration de Lutathera [voir Avertissements et précautions ].
Populations spécialesPatients pédiatriques
Il n'y avait pas de différences cliniquement pertinentes dans l'exposition du lutetium Lu 177 dotatate chez les patients pédiatriques de 12 ans et plus par rapport à celui des patients adultes.
Études d'interaction médicamenteuseÉtudes in vitro
Enzymes CYP450
Le lutetium non radioactif Lu 175 dotatate n'est pas un inhibiteur ou un inducteur du cytochrome P450 (CYP) 1A2 2B6 2C9 2C19 ou 2D6 in vitro .
Transporteurs
Le lutetium LUTEITIA LU 175 non radioactif n'est pas un inhibiteur de la glycoprotéine P BCRP OAT1 OAT3 OCT1 OCT2 OATP1B1 ou OATP1B3 in vitro .
Toxicologie et / ou pharmacologie animale
Le principal organe cible dans les études animales utilisant le lutetium Lutetium LU 175 non radioactif dotaTate était le pancréas un organe exprimant SSTR2 élevé. L'apoptose acineuse pancréatique s'est produite à des doses dotatées lutétium LU 175 ≥ 5 mg / kg dans des études de toxicologie à dose répétée chez le rat. Une atrophie des cellules acinaires pancréatiques s'est également produite dans des études de toxicologie à dose répétées chez des chiens à des doses ≥ 500 mcg / kg. Ces résultats étaient cohérents avec une forte absorption du peptide radiomarqué dans le pancréas dans les études de biodistribution animale.
Études cliniquesTumeurs carcinoïdes progressifs avancées ou métastatiques progressives
L'efficacité de la Lutathera chez les patients présentant des tumeurs carcinoïdes moyennes et métastatiques de la somatostatine à la somatostatine intermédiaire de la somatostatine intermédiaire de la somatostatine localement avancées / inopérables ou métastatiques a été établie dans Netter-1 (NCT01578239) Un essai à complage actif à l'absence à contrôle randomisé multicentrique. Les critères d'éligibilité clés comprenaient l'indice KI67 ≤ 20% de la performance de Karnofsky ≥ 60 présence confirmée de récepteurs de la somatostatine sur toutes les lésions (absorption d'Octreoscan ≥ Liver normal) Créinine Créatine ≥ 50 ml / min Aucun traitement antérieur avec la thérapie radiothérapie du récepteur de la peptide (PRRT) et pas de radiothérapie externe antérieure pour plus de mer.
Au moment de l'analyse primaire, 229 patients ont été randomisés (1: 1) pour recevoir soit Lutathera 7,4 GBQ (200 MCI) toutes les 8 semaines (± 1 semaine) pour jusqu'à 4 administrations (dose cumulative maximale de 29,6 gbq) ou l'octréotide à longue durée à action élevée (défini comme 60 mg par injonction intramusculaire toutes les 4 semaines). Les patients du bras Lutathera ont également reçu un octreotide à longue durée d'action 30 mg comme injection intramusculaire 4 à 24 heures après chaque dose de Lutathera et toutes les 4 semaines après la fin du traitement de Lutathera jusqu'à la progression de la maladie ou jusqu'à la semaine 76 de l'étude. Les patients des deux armes pourraient recevoir un octréotide à courte durée d'action pour la gestion des symptômes; Cependant, l'octréotide à action courte a été retenu au moins 24 heures avant chaque dose de Lutathera. La randomisation a été stratifiée par le score d'absorption de la tumeur Octreoscan (grade 2 3 ou 4) et la durée où les patients avaient été sur la dose constante la plus récente d'octréotide avant la randomisation (≤ 6 ou> 6 mois). La principale mesure des résultats de l'efficacité était la survie sans progression (PFS), telle que déterminée par un comité d'examen indépendant (IRC) en aveugle selon RECIST v1.1. Des mesures supplémentaires sur les résultats de l'efficacité ont été le taux de réponse global (ORR) par la durée de la réponse de l'IRC (DOR) par IRC et la survie globale (OS).
Les caractéristiques démographiques et de base de la maladie étaient équilibrées entre les bras de traitement. Sur les 229 patients, 82% étaient blancs 4% étaient noirs 3% étaient hispaniques ou latinos 0,4% étaient asiatiques 0,4% étaient d'autres et 9% n'étaient pas signalés. L'âge médian était de 64 ans (28 à 87 ans); 51% étaient des hommes, 74% avaient une primaire iléale et 96% avaient une maladie métastatique dans le foie. Le score de performance médian de Karnofsky était de 90 (60 à 100), 74% ont reçu une dose constante d'octréotide pendant> 6 mois et 12% ont reçu un traitement antérieur avec l'évérolimus. Soixante-neuf pour cent des patients avaient une expression de Ki67 dans ≤ 2% des cellules tumorales 77% avaient un CGA> 2 fois la limite supérieure de 65% normal (ULN) avait 5-HIAA> 2 fois ULN et 65% avaient une phosphatase alcaline ≤ ULN.
Au moment de l'analyse finale du SG qui s'est produite 66 mois après l'analyse PFS primaire, 117 patients ont été randomisés pour le bras de Lutathera et 114 patients ont été randomisés pour le bras octreotide. Dans l'analyse finale du système d'exploitation, il n'y avait pas de différence statistiquement significative dans la SG entre les deux bras de traitement.
Les résultats de l'efficacité pour Netter-1 sont présentés dans le tableau 9 et la figure 1.
Tableau 9. L'efficacité se traduit par Netter-1
| Lutathera with Long-acting Octreotide (30 mg) N = 116 | Octreotide à action prolongée (60 mg) N = 113 | |
| PFS par IRC | ||
| 27 (23%) | 78 (69%) | |
| 15 (13%) | 61 (54%) | |
| 12 (10%) | 17 (15%) | |
| Non (18.4 non) | 8.5 (6.0 9.1) | |
| a (95% là-bas) | 0,21 (0,13 0,32) | |
| b | <0.0001 | |
| Orr par IRC | ||
| 13% (7% 19%) | 4% (0,1% 7%) | |
| 1 (1%) | 0 | |
| 14 (12%) | 4 (4%) | |
| c | 0.0148 | |
| Non (2.8 non) | 1.9 (1.9 NE) | |
| Abréviations: intervalle de confiance CI; IRC independent radiology committee; NON pas évaluable; NR n'est pas atteint; Taux de réponse global de l'ORR; Survie sans progression PFS. aRatio de risque basé sur le modèle COX non structifié. bTest de classement des journaux non stratifiés. cTest exact de Fisher. |
Figure 1. Courbes de Kaplan-Meier pour la survie sans progression dans Netter-1
Tumeurs neuroendocrines gastroentéroprantes pour les récepteurs de la somatostatine positifs
L'efficacité de la Lutathera chez les patients souffrant d'intestin moyen et de gastroentéropancréopancréatique neuroendocrine (réseau GEP) a été évaluée chez 360 patients de l'étude Erasmus. Dans Erasmus Lutathera, a été initialement fourni comme un accès étendu sous un protocole général de thérapie par radionucléide des récepteurs peptidiques sur un seul site aux Pays-Bas. Un protocole spécifique à Lutathera ultérieur écrit huit ans après l'initiation de l'étude n'a pas décrit un plan de test de taille ou d'hypothèse spécifique, mais a permis une collecte de données rétrospective. Au total, 1214 patients ont reçu Lutathera chez Erasmus dont 578 patients avaient des évaluations tumorales de base. Sur les 578 patients, 360 (62%) avaient des réseau GEP et un suivi à long terme. Parmi ces 360 patients 145 (40%) ont eu leurs tumeurs évaluées de manière prospective selon les critères RECIST. Lutathera 7,4 GBQ (200 MCI) a été administré toutes les 6 à 13 semaines jusqu'à 4 doses en même temps que la solution d'acides aminés recommandée. Le principal résultat de l'efficacité était l'ORR évalué par l'enquêteur. L'âge médian dans le sous-ensemble d'efficacité était de 60 ans (30 à 85 ans) 51% étaient des hommes de 71% avaient un statut de performance de Karnofsky de base ≥ 90 51% avaient progressé dans les 12 mois suivant le traitement et 7% avaient reçu avant chimiothérapie . Cinquante-deux pour cent (52%) des patients ont reçu un analogue concomitant de la somatostatine. La dose médiane de Lutathera était de 29,6 GBQ (800 MCI). L'ORR évalué par les chercheurs était de 17% (IC à 95%: 13 21) sur la base d'une analyse qui obligeait les intervenants à avoir subi des évaluations de réponse prospectives selon les critères RECIST. Trois réponses complètes ont été observées (<1%). Median DoR in the 60 responding patients was 35 months (95% CI: 17 38).
Informations sur les patients pour Lutathera
Risque de l'exposition aux radiations
Conseiller les patients et / ou les soignants pour minimiser l'exposition aux radiations aux contacts des ménages pendant et après le traitement avec Lutathera conformément aux bonnes pratiques institutionnelles de radiothérapie et des procédures de gestion des patients [voir Posologie et administration Avertissements et précautions ].
Myélosuppression
Conseiller aux patients et / ou aux soignants de contacter leur fournisseur de soins de santé pour tout signe ou symptôme de myélosuppression ou d'infection [voir Avertissements et précautions ].
Syndrome myélodysplasique secondaire et leucémie
Conseiller les patients et / ou les soignants du potentiel de cancers secondaires, y compris le syndrome myélodysplasique et la leucémie aiguë [voir Avertissements et précautions ].
Toxicité rénale
Conseiller les patients et / ou les soignants pour hydrater et uriner fréquemment avant le jour de et le lendemain de l'administration de Lutathera [voir Avertissements et précautions ]. Advise patients to contact their healthcare provider for any signs or symptoms of renal toxicity [voir Avertissements et précautions ].
Hépatotoxicité
Conseiller les patients et / ou les soignants de la nécessité de tests de laboratoire périodiques pour surveiller l'hépatotoxicité [voir Avertissements et précautions ]. Advise patients to contact their healthcare provider for any signs or symptoms of hepatotoxicity [voir Avertissements et précautions ].
Hypersensibilité
Informer les patients et / ou les soignants que Lutathera peut provoquer des réactions d'hypersensibilité, y compris l'œdème de l'angio Avertissements et précautions ].
Crises hormonales neuroendocrines
Conseiller aux patients et / ou aux soignants de contacter leur fournisseur de soins de santé pour des signes ou des symptômes qui peuvent survenir après une libération hormonale liée à la tumeur [voir Avertissements et précautions ].
Toxicité embryo-fœtale
Conseiller les femmes enceintes et les hommes et les femmes du potentiel reproducteur du risque potentiel pour un fœtus. Conseiller aux femmes d'informer leur fournisseur de soins de santé d'une grossesse connue ou suspectée [voir Avertissements et précautions Utiliser dans des populations spécifiques ].
Conseiller les femmes de potentiel reproducteur à utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 7 mois après la dernière dose [voir Utiliser dans des populations spécifiques ].
Conseiller les patients masculins avec des partenaires féminins de potentiel reproducteur pour utiliser une contraception efficace pendant le traitement avec Lutathera et pendant 4 mois après la dernière dose [voir Utiliser dans des populations spécifiques ].
Lactation
Conseiller les femmes de ne pas allaiter pendant le traitement avec Lutathera et pendant 2,5 mois après la dernière dose [voir Utiliser dans des populations spécifiques ].
Infertilité
Conseiller les patientes féminines et masculines que Lutathera peut nuire à la fertilité [voir Avertissements et précautions Utiliser dans des populations spécifiques ].